8—羧基喹唑啉酮衍生物的合成
2017-05-30 10:48:04徐玉玲何汀陳晶晶羅珊珊
安徽農業科學 2017年8期
徐玉玲 何汀 陳晶晶 羅珊珊

摘要以2-氨基-3-甲基苯甲酸為起始原料,經醋酸酐乙酰化、氧化、關環、取代等反應,設計合成了一系列8-羧基-2-甲基-3-芳基(脂肪基)-4(3H)-喹唑啉酮衍生物,所有目標化合物均測定了熔點并通過了1H NMR和質譜結構表征。通過單晶結構測試,發現該類化合物存在很強的氫鍵,很難進一步酯化或合成酰胺。
關鍵詞喹唑啉酮;氧化;衍生物
中圖分類號O626文獻標識碼A文章編號0517-6611(2017)08-0001-03
Synthesis of 8Carboxyquinazolinone Derivatives
XU Yuling, HE Ting, CHEN Jingjing et al
(School of Chemical & Environmental Engineering, Wuhan Polytechnic University, Wuhan, Hubei 430023)
AbstractThe synthesize route of 2methyl4oxo3phenyl3,4dihydroquinazoline8carboxylic acid derivatives undergoed acetylation, oxidation, cyclization and substitution reactions from the starting material of 2amino3methylbenzoic acid. All of the target compounds were confirmed by 1HNMR, MS and melting point. According to the result of Xray diffraction analysis, this series compounds existed strong hydrogen bond that was very difficult for further reaction of esterification or amidation.
Key wordsQuinazolinone;Oxidation;Derivatives
喹唑啉酮類化合物因其結構的可變性和廣泛的生物活性,且合成方法多種多樣,是雜環衍生物中重要的一類化合物,在農藥和醫藥領域有著廣泛應用[1-4]。在農藥領域,喹唑啉酮的功效主要表現在殺蟲、殺菌、抗病毒等方面;在醫藥領域,喹唑啉酮的功效主要表現在抗菌、抗腫瘤、抗癌、抗HIV-l和抗結核等方面,而且還具有退熱、抗炎、催眠、抗驚厥、抗帕金森綜合征、調節心血管和調節細胞及酶的活性功能。近年來,喹唑啉酮類化合物是有機合成、醫藥、農藥和其他精細化工領域研究開發的熱點課題之一[5-8]。
喹唑啉酮類化合物具有分子量小、結構簡單、活性譜廣等優點。設計合成的喹唑啉酮衍生物,不少都保留了喹唑啉酮的優點,活性也有很大的提高。因此,以喹唑啉酮為先導化合物,設計、合成結構新穎的化合物,并對其進行生物活性研究,具有很高的藥物開發價值[9-10]。鑒于此,筆者以2-氨基-3-甲基苯甲酸為原料,經過乙酰化、氧化、關環、取代等反應,設計合成了一系列8-羧基喹唑啉酮衍生物,以期篩
選出具有進一步修飾潛力的醫藥先導化合物。
1材料與方法
1.1材料
1H NMR和13C NMR譜采用Mercury Plus 400 MHz核磁共振儀或者Varian VNMR 600 MHz(TMS為內標,DMSO-D6為溶劑);質譜ESI-MS由API2000質譜儀測定;元素分析數據由VarioELⅢ CHNSO型元素分析儀測定;單晶衍射用Bruker Smart Apex CCD型X-ray單晶衍射儀;熔點由BüCHI B-545數字熔點儀(溫度計未經校正)測定。德國Heidolph MR3001型恒溫加熱磁力攪拌器,瑞士BüCHI R-200型旋轉蒸發儀、德國Sartoruis電子天平、ZF-20D暗箱式紫外分析儀、DLSB低溫冷卻循環泵、SHZ-D(Ⅲ)循環水式真空泵、70-1型遠紅外線干燥箱、點樣硅膠板等儀器。試劑和溶劑如無特殊說明,均為分析純或化學純。
1.2方法
1.2.1化合物1~3的合成。
以簡單易得的2-氨基-3-甲基苯甲酸為起始原料,經醋酸酐乙酰化、氧化、關環、取代等反應,設計合成了一系列8-羧基-喹唑啉酮衍生物,具體合成路線見圖1。
1.2.1.1 化合物1的合成。
15.100 g (100 mmol)2-氨基-3-甲基苯甲酸與40 mL乙酸酐于100 mL圓底燒瓶中,回流4.5 h至反應完全,停止加熱,立即將熱溶液(褐色)倒入350 mL冰水中,溶液中出現白色絮狀沉淀,繼續攪拌10 h后,過濾,干燥,得白色固體16.500 g,收率為86%。
1.2.1.2化合物2的合成。
將9.660 g (50 mmol)化合物1溶于200 mL水中,升溫至80 ℃,分批緩慢加入31.600 g KMnO4,待紫色消失后繼續攪拌6 h,反應結束后溶液呈灰褐色,趁熱過濾,上層濾渣用熱水洗2次,得淺黃色濾液,冷卻后,濾液用濃鹽酸酸化,析出白色沉淀,過濾,干燥得白色固體7.700 g,產率為70%。
1.2.1.3化合物3的合成[9]。
將8.927 g (40 mmol)化合物2與40 mL乙酸酐置于100 mL圓底燒瓶中,加熱攪拌回流4 h至反應完全,減壓蒸餾除去乙酸酐。加入正己烷重結晶,得黃色固體,過濾,用石油醚洗2次,正己烷洗2次,干燥得淡黃色固體(化合物3)6.800 g,收率為83%。熔點為211~212 ℃。
1.2.2目標化合物4的合成[9]。
1.2.2.1化合物4a~4h的合成。
在50 mL圓底燒瓶中加入2.050 g (10 mmol)化合物3和10 mmol取代苯胺化合物,室溫下攪拌3 h左右,加入50 mL 10%NaOH溶液,繼續攪拌4 h,然后用濃鹽酸酸化至溶液pH為1~2,靜置,過濾,上層固體用水洗3次,石油醚洗1次,干燥得粗產品,用無水乙醇重結晶,干燥得目標產物4a~4h。
1.2.2.2化合物4i~4j的合成。
在50 mL圓底燒瓶中加入0.620 g (3 mmol)化合物3和3 mmol脂肪胺類化合物,加熱回流24 h后,加入20 mL水,然后用濃鹽酸酸化至溶液pH為1~2,有沉淀析出,靜置,過濾,上層固體用水洗2次,石油醚洗1次,干燥得粗產品,用無水乙醇重結晶,干燥得目標產物4i~4j。
2結果與分析
2.1目標化合物的性質
芳胺取代喹唑啉酮化合物見圖2和表1,脂肪胺取代喹唑啉酮化合物見圖3和表2。所有化合物收率均為中等,這是因為得到目標化合物后,采用乙醇進行重結晶,損失一部分產品。在相同的后處理條件下,當苯環上的取代基為吸電子基團如氯原子時,產物收率偏低,當取代基為雙取代時如4g和4h,收率也偏低。這可能是因為苯環上為吸電子基或者雙取代基時反應較難進行,所以收率略微偏低。當R取代基為烷基如乙基或者丙基時,合成方法與苯環取代基完全不同,需要改為加熱回流狀態,且酸化時改為鹽酸,使未反應的烷基胺形成銨鹽,便于去除。
2.2目標化合物的進一步衍生
為了解目標化合物4的衍生能力及作為醫藥中間體的前景,該研究將化合物4進行進一步的衍生(以4b為例),使用酰化試劑將其轉變為酰氯,以便下一步合成酯或者酰胺,并進行了二氯亞砜、草酰氯、三氯氧磷、三氯化磷、五氯化磷等與4b反應,均未成功得到相應的酰氯。合成酰胺或者酯的另一種方法是使用縮合劑,根據文獻[11-12]報道,該研究使用的縮合劑有DCC/DMAP、EDCI/DMAP、DIC/DMAP、HOBT/EDCI、NHS/EDCI、HOOBT/EDCI等,使用這些縮合劑反應3 d后仍然未檢測到新點。
所嘗試的酰氯法、縮合法及微波法均未得到目標酯或者酰胺,分析無法得到目標物的原因可能有以下幾個方面:①生成酯的位阻比較大,用一般的方法無法完成。②酸的羥基和鄰位上的氮形成很強的氫鍵,使得酰氯難以形成從而難以合成相應的酯。③苯環上有吸電子取代基時,酰氯反應較難進行。因此,繼續對化合物4b的結構進行研究。
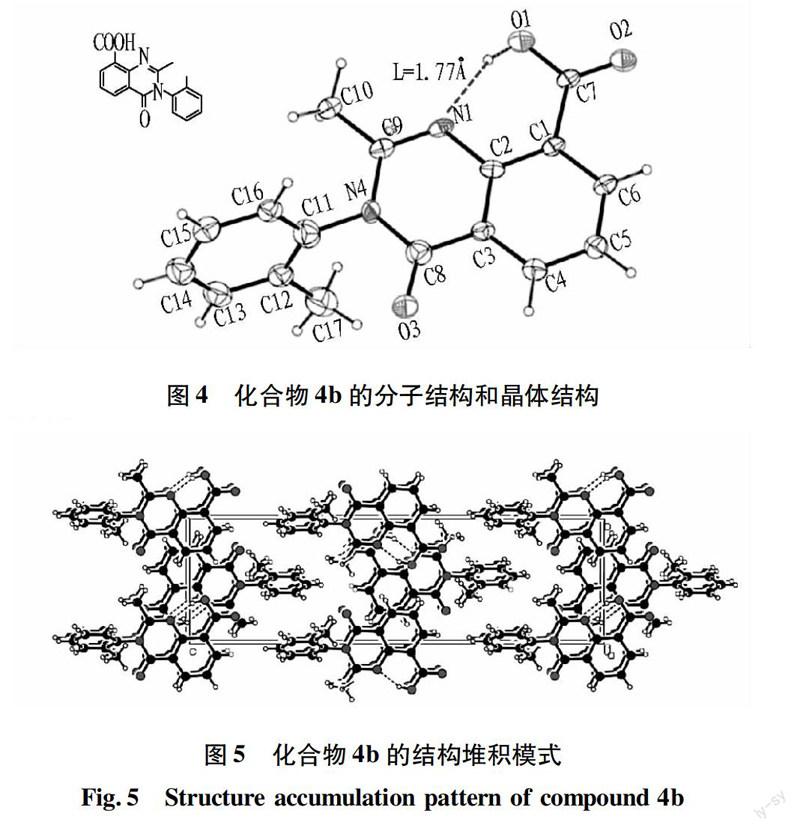
2.3化合物4b的單晶衍射分析
X-ray單晶衍射試驗在Bruker Smart Apex CCD型X-ray單晶衍射儀上進行,采用經石墨單色化的Mo-Kα射線為入射光,波長為0.710 73 。衍射試驗是在T=298 K下進行的。選擇尺寸合適的單晶進行衍射試驗。結構用直接法結合Fourier合成解出。所有原子坐標均由理論加氫確定,修正采用“騎”母原子模式精修。
根據IUPAC給出的氫鍵準則:分子內氫鍵必須具備形成氫鍵的必要條件,還要具有特定的條件,例如:形成平面環,環的大小以五或六元環最穩定,形成的環中沒有任何的扭曲。O—H……N之間的二面角是直線或接近180°。氫鍵越強,H……N距離越短。由圖4可知,化合物4b的單晶衍射顯示分子內形成很強的氫鍵,N1與O1上氫原子間距離僅為1.77 。N1和O1上氫原子幾乎形成一條直線,由N1、CI、C2、C7、O1及其氫原子等形成穩定的平面五元環結構,所以整個分子具有很強的分子內氫鍵,這也是這一類化合物難以進行酯化或者酰化反應的一個重要原因。
e of compound 4b
氫鍵在成鍵方向的最優選擇影響晶體的結構堆積模式。為化合物4b的結構堆積模式,可見整個喹唑啉酮結構為一個平面,由氮原子連接的鄰甲基苯環與這個平面形成一個夾角為77°的二面角。因此,分子的堆積模式為平面層狀結構。
3結論
以簡單易得的2-氨基-3-甲基苯甲酸為起始原料設計合成了一系列8-羧基-2-甲基-3-芳基(脂肪基)-4(3H)-喹唑啉酮衍生物,所有目標化合物均測定了熔點并通過了1H NMR和質譜結構表征。
通過單晶結構測試,發現該類化合物存在很強的分子內氫鍵,所以很難進一步合成酯或酰胺。
參考文獻
[1] ELBAYOUKI K A H M,BASYOUNI W M,MOHAMED Y A F,et al.Novel 4(3H)quinazolinones containing biologically active thiazole,pyridinone and chromene of expected antitumor and antifungal activities[J].Euro J Chem,2011,2(4):455-462.
[2] XU Y L,LIN H Y,CAO R J,et al.Pyrazolonequinazolone hybrids:A novel class of human 4hydroxyphenylpyruvate dioxygenase inhibitors[J].Bioorg Med Chem,2014,22(19):5194-5211.
[3] AVALLONE A,DIGENNARO E,SILVESTRO L,et al.Targeting thymidylate synthase in colorectal cancer:Critical reevaluation and emerging therapeutic role of raltitrexed[J].Expert Opin Drug Saf,2014,13(1):113-129.
[4] RIVERO I A,ESPINOZA K,SOMANATHAN R.Syntheses of quinazoline
2,4dione alkaloids and analogues from Mexican Zanthoxylum species[J].Molecules,2004,9(7):609-616.
[5] BOYLES D C,CURRAN T T,PARLETT R V.Electrophilic Namination of two quinazoline2,4diones using substituted(nitrophenyl)hydroxylamines[J].Org Process Res Dev,2002,6(3):230-233.
[6] LI J R,CHEN X,SHI D X,et al.A new and facile synthesis of quinazoline2,4(1H,3H)diones[J].Org Lett,2009,11(6):1193-1196.
[7] LI Y X,LUO Y P,XI Z,et al.Design and syntheses of novel phthalazin1(2H)one derivatives as acetohydroxyacid synthase inhibitors[J].J Agric Food Chem, 2006,54(24):9135-9139.
[8] ZAYED M F,HASSAN M H.Synthesis and biological evaluation studies of novel quinazolinone derivatives as antibacterial and antiinflammatory agents[J].Saudi Pharm J,2014,22(2):157-162.
[9] 羅鐵軍,李正名,趙衛光,等.2-甲基-3-芳基-7-(5,5-二甲基-3-酮-1-環己烯-1-基)甲酸酯-4(3H)-喹唑啉酮的合成[J].有機化學,2002,22(10):741-745.
[10] TRIPATHY,PRADEEP K.Microwave irradiated one flask synthesisof 2,3disubstituted quinazolin4ones[J].Journal of the institution of chemists,2003,75(6):179-180.
[11] MALHOTRA S,KOUL S K,SHARMA R L,et al.Studies on some biologically active azepinoquinazolines:Part I.An approach to potent bronchodilatory compounds[J].Organic chemistry including medicinal chemistry,1988,27(10):937-940.
[12] ELFAHAM A,ALBERICIO F.Peptide coupling reagents,more than a letter soup[J].Chem Rev,2011,111(11):6557-6602.
