超級電容器及鋰電池中鋰離子相互作用模型的構建
2017-02-28 08:30:46劉宇江浩李春忠劉洪來
化工學報 2017年2期
關鍵詞:模型
劉宇,江浩,李春忠,劉洪來
(1化學工程聯(lián)合國家重點實驗室,華東理工大學化工學院,上海 200237;2華東理工大學材料科學與工程學院,超細粉末教育部重點實驗室,上海 200237;3化學工程聯(lián)合國家重點實驗室,華東理工大學化學與分子工程學院,上海 200237)
超級電容器及鋰電池中鋰離子相互作用模型的構建
劉宇1,江浩2,李春忠2,劉洪來3
(1化學工程聯(lián)合國家重點實驗室,華東理工大學化工學院,上海 200237;2華東理工大學材料科學與工程學院,超細粉末教育部重點實驗室,上海 200237;3化學工程聯(lián)合國家重點實驗室,華東理工大學化學與分子工程學院,上海 200237)
超級電容器及鋰電池中鋰離子高度富集,構建準確的鋰離子相互作用模型對于預測超級電容器及鋰電池性能、設計電極材料具有重要的指導作用。通過量子密度泛函理論計算了超級電容器及鋰電池中鋰離子間的相互作用,重點考察了鋰離子間短程范德華相互作用的特點及溶劑化效應對范德華作用的影響,發(fā)現(xiàn)短程區(qū)域內范德華作用能在很大程度上屏蔽庫侖排斥作用,溶劑化效應對范德華作用有很大貢獻。通過數(shù)值擬合建立了能適用于不同溶劑環(huán)境下的鋰離子相互作用分子模型(隱式溶劑模型)。另外還考察了鋰離子間三體相互作用,發(fā)現(xiàn)三體相互作用為吸引作用,且僅對局部大量富集的鋰離子有較大影響。
鋰離子;超級電容器;密度泛函理論;模型;溶劑;微尺度
引 言
鋰離子超級電容器和鋰電池具有能量密度高、穩(wěn)定性好、材料制備簡單、綠色無污染等諸多優(yōu)良特性,具有廣闊的應用前景,是當前化工、能源、環(huán)境等領域的研究熱點[1-11]。Cao等[12]制備了MoS2/石墨烯復合的鋰電池材料,該鋰離子電池以EC/DEC混合溶液為電解液,儲能密度可達877 mA·h·g-1,并能在前50步充放電循環(huán)中保持穩(wěn)定。Wang等[13]以TiC納米顆粒鏈作為陽極,以吡啶衍生的分層摻氮碳材料為陰極,合成了TiC/PHPNC鋰離子超級電容器,該電容器具有101.5 W·h·kg-1的儲能密度和67.5 kW·kg-1的功率密度,并在5000次充放電循環(huán)中保持有82%的初始效果。最近,Jiang等[14]通過在碳納米管中摻入納米銀制備了復合型鋰電池材料,該材料以EC/DMC混合溶液作為電解液,將儲能密度提高到1637 mA·h·g-1并成功抑制了充放電過程中SEI膜的形成。由于鋰離子超級電容器和鋰電池的高性能、高穩(wěn)定性,目前已有大量廠商開始制造生產(chǎn)鋰離子電池和研發(fā)鋰離子超級電容器。
鋰離子間的相互作用對電容器和鋰電材料的性能具有決定性作用,深入認識鋰離子間相互作用,構建準確的鋰離子分子模型對于預測超級電容器性能、設計電極材料具有重要的指導作用。目前,對鋰離子間相互作用模型通常是在短程范德華勢(如Lennard-Jones勢、硬球勢等)的基礎上加長程庫侖相互作用。其中范德華勢中的分子參數(shù)一般通過擬合實驗數(shù)據(jù)(如溶解自由能等)[15-16]或直接由標準力場給定[17-18]。但擬合分子參數(shù)所用的實驗數(shù)據(jù)往往是由鋰離子濃度較低的溶液測得,與鋰離子高度富集的超級電容器和鋰電池的實際情況有較大區(qū)別;標準力場中的鋰原子參數(shù)往往針對化合態(tài)而非游離態(tài)的鋰原子,也不能完全反映超級電容器和電池材料中鋰離子的特征。由于靜電屏蔽效應,溶劑化效應對鋰離子間相互作用有重要影響,因此,在隱式溶劑模型中,鋰離子的分子參數(shù)原則上應當與溶劑的相對介電常數(shù)相關。目前常用的鋰離子的分子模型往往針對真空或水溶液中的鋰離子;而在超級電容器和鋰電池中,鋰離子經(jīng)常存在于非水溶液中。目前常用的非水系溶液包括有機溶劑(如EC/DEC[19]、EC/DMC[14])、離子液體(如TFSI/BF4[20])等,其熱力學、動力學性質與水溶液均有顯著差異。即使對于水系溶液,由于電解質(H2SO4、KOH等)的存在,其電化學性質亦與純水溶液有較大不同。因此,要準確預測鋰離子在超級電容器和電池材料中的行為特征就必須建立能適用于不同溶劑的鋰離子分子模型。
理論上,基于量子力學第一性原理建立分子相互作用模型是最為嚴謹?shù)淖龇╗21-25]。例如Joung等[26]結合量化計算、分子模擬以及實驗擬合建立了水溶液中針對不同水分子模型(SPC、SPC/E、TIP3P等)的堿金屬離子和鹵素離子的分子力場。Pezeshki等[27]采用柔性邊界量子力學/分子力學(FB-QM/MM)方法考察了Li+、Na+、NH+4和Cl-在水溶液中的電荷分布情況,并發(fā)現(xiàn)需要在分子力學(MM)的力場參數(shù)中引入經(jīng)驗性修正才能得到合理的結果,他們的工作證明了即使在顯式溶劑模型中,分子參數(shù)依然受到溶劑化效應的影響,建立與溶劑相關的分子力場參數(shù)能顯著改善理論預測結果。本文采用量子力學的密度泛函理論分析鋰離子在不同溶劑中的相互作用,考察鋰離子間的多體相關性,建立適用于不同溶劑的鋰離子分子模型。
1 模型與理論
1.1 分子間勢能模型
按經(jīng)典力學的假設,含有N個粒子的系統(tǒng)的總能量E為粒子間相互作用的對加和
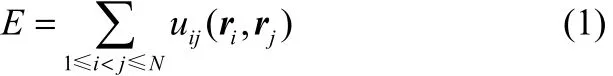
其中,uij(ri,rj)為第i與第j個粒子間相互作用勢能函數(shù);ri、rj分別為第i和第j個粒子的坐標。要建立鋰離子的分子模型,實際上就是給出uij(ri,rj)的具體形式。從量子力學的角度而言,系統(tǒng)的總能量除了包含對加和貢獻外還包含多體相關貢獻,因而不能直接寫為式(1)的形式;根據(jù)貢獻的強弱,E可以展開為
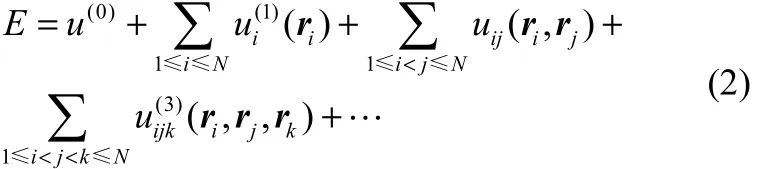
式中,u(0)與零勢點的選取有關,為孤立的單個粒子所具有的能量,u(i)為i體相關能。對于無外場的系統(tǒng),單個粒子自能ui(1)與位置無關,可通過定義零勢點消去;如果忽略高階相關項的貢獻,式(2)即可回歸到經(jīng)典力學的式(1)。
為了得到uij(ri,rj)的具體形式,考慮僅含兩個鋰離子的體系,N= 2,并選取u(0)=0,式(2)可寫為

通過量子力學理論計算式(3)中的總能量E以及自能即可建立鋰離子的分子模型。
1.2 量子密度泛函理論
對于含有M個電子的系統(tǒng),其能量本征值E原則上可由多體薛定諤方程(原子單位制)
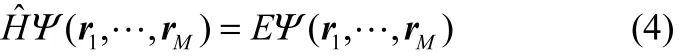
求解,其中

其中,V(r1,…,rM)為外部勢場,一般由原子核-電子相互作用貢獻。由于Ψ(r1,…,rM)為3M維函數(shù),直接求解式(4)是非常耗時的。為此,Kohn等[28-29]提出了以3維的電子云密度n(r)取代3M維波函數(shù)的Ψ(r1,…,rM)的Kohn-Sham密度泛函理論(Kohn-Sham density functional theory, KS-DFT)。
在KS-DFT中,電子云密度n(r)被定義為
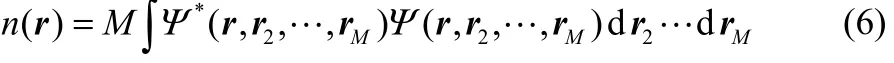
式(6)表明,電子云密度n(r)是多體波函數(shù)Ψ(r1,…,rM)的唯一泛函;進一步,Kohn等[28-29]還證明了多體波函數(shù)Ψ(r1,…,rM)亦是電子云密度n(r)的唯一泛函,Ψ[n]。因此,系統(tǒng)內能E也可表示為電子云密度n(r)的唯一泛函

根據(jù)式(5),E可進一步被分解為動能項T,外勢場項V,電子間相互作用項J以及交換相關項Exc

在KS-DFT中,動能項T被隱式地表示為n(r)的泛函
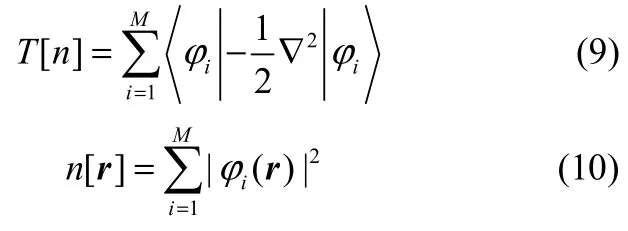
其中,φi(r)為單電子軌道波函數(shù)。如果不引入φi(r)而將T顯式地表示為n(r)的泛函,則為無軌道密度泛函理論(orbital-free density functional theory, OF-DFT)[30-32]。由于OF-DFT中對動能項引入了近似,因而精度較KS-DFT低,在精度要求較高的系統(tǒng)中(如本文所討論的鋰離子分子相互作用模型),一般采用KS-DFT。
外勢場、電子間相互作用項均可精確表示為電子云密度的泛函
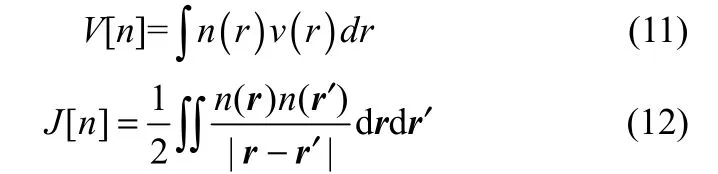
其中,v(r)為單電子所受的外部勢場。
交換相關能Exc源于電子的不可分辨性(交換能Ex)以及動能-靜電相關效應(相關能Ec),可由絕熱連接(adiabatic connection)方程計算[33]
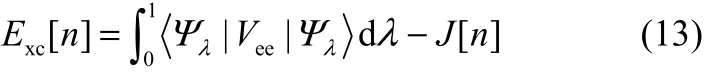
但一般不能解析地表示為電子云密度的泛函。圍繞交換相關能,人們提出了大量的近似理論,如局域密度近似(local density approximation, LDA)[34-35]、加權密度近似(weighted density approximation,WDA)[36-38]、PBE近似[39]、B3LYP近似[40-41]等。本文將采用B3LYP近似。
結合式(8)~式(12),即可建立內能關于電子云密度的泛函形式。通過能量極小化,即可得到定態(tài)電子云密度分布以及內能本征值。
本文的量子力學計算通過Material Studio 軟件的Dmol3模塊完成,交換相關能采用B3LYP近似,單電子波函數(shù)通過DNP3.5基組展開,收斂精度為2.623×10-6kJ·mol-1,溶劑化效應采用COSMO模塊計算。
2 結果與討論
2.1 鋰離子兩體相互作用
首先考慮鋰離子間兩體相互作用,如圖1所示。通過計算不同r值對應的系統(tǒng)內能構建u(r)的具體形式。對于離子間相互作用,其勢能函數(shù)一般可拆分為長程靜電相互作用uele(r)和短程范德華相互作用uvdw(r)。其中長程項可由庫侖定律精確得到

其中,ε0為真空中的介電常數(shù),εr為溶劑的相對介電常數(shù),zi為粒子i所帶電荷數(shù),e為單位電荷的電量。在總勢能函數(shù)中扣除長程項即可得到鋰離子間的范德華相互作用勢
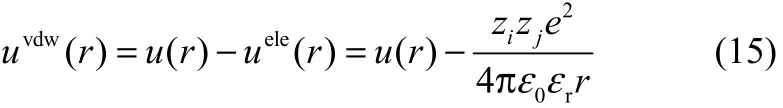

圖1 鋰離子兩體相互作用示意圖Fig.1 Diagrammatic sketch of two body interaction between lithium ions
圖2為真空中鋰離子間的相互作用勢。其中總相互作用勢(total)由量化計算[式(3)]得到,靜電勢(ele)由式(14)計算,范德華勢(vdw)由式(15)計算。雖然鋰離子間總相互作用為排斥,但當0.1 nm<r<0.5 nm時,排斥勢能明顯低于庫侖相互作用所貢獻的排斥勢,而范德華相互作用在r>r0= 0.058 nm時由排斥轉為吸引;而當r=rmin= 0.091 nm時達到最低值(勢阱)uvdw(r) = -εmin;勢阱值 -1138.2 kJ·mol-1遠低于一般分子相互作用模型中的勢阱值。這也就意味著在短程區(qū)間內,范德華相互作用可以中和大部分的庫侖排斥作用;而這種中和效應,很難通過常用的LJ-12-6勢能模型正確反映。為此,在0.1 nm<r<0.5 nm區(qū)間內采用LJ-m-n勢能擬合
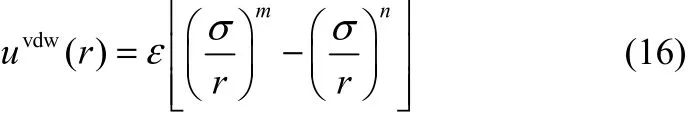
發(fā)現(xiàn)當m= 4,n= 1時效果最佳。值得注意的是式(16)僅適用于短程相互作用,不能直接推廣到長程相互作用。在長程區(qū)域(r>2 nm),uvdw(r)按r-5.81衰減,uvdw(r)-(1.42 nm/r)-5.81,因此不能通過式(16)計算。長程相互作用以5.81次方衰減的結果與色散力的長程六次方衰減的結論基本一致;此時,范德華相互作用的貢獻遠低于靜電貢獻,分子間相互作用由庫侖勢主導。原則上可以通過采用比式(16)更復雜的函數(shù)形式和引入更多的分子參數(shù)建立適用于全區(qū)間的鋰離子范德華模型,但考慮到實際應用中(如分子模擬)勢能函數(shù)應具有簡潔的函數(shù)形式,而且長程區(qū)范德華作用較弱,范德華截斷半徑一般在3~5個分子直徑之間,因此,本文僅對短程相互作用進行LJ-m-n擬合。
圖3所展示的為6種不同溶劑(DEC—碳酸二乙酯;MF—三聚氰胺甲醛樹脂;ET—乙醇;GBL—γ-丁內酯;water—水;EC—碳酸乙烯酯)中鋰離子間相互作用勢(對于未列出的溶劑,可通過對介電常數(shù)進行插值估算;對于混合溶劑,介電常數(shù)可由立方根相加率計算)。與真空中相互作用類似,短程區(qū)間內,范德華相互作用對靜電排斥勢有顯著的中和效應。不同溶劑中鋰離子的范德華相互作用呈現(xiàn)出明顯的差異,隨著相對介電常數(shù)εr的增大,范德華作用的零點能坐標r0以及勢阱坐標rmin呈現(xiàn)明顯的增大趨勢,而勢阱深度εmin則呈現(xiàn)明顯的減小趨勢(具體數(shù)值見表1)。其變化趨勢如圖4所示,r0與rmin在εr∈[1,20]區(qū)間內呈現(xiàn)急劇的上升趨勢,變化幅度為0.05~0.45 nm,而當εr> 20時,變化則較為平緩;相對地,阱深εmin則在εr< 10時,迅速地由1138 kJ·mol-1衰減到17 kJ·mol-1,而當εr> 20時,變化同樣較為平緩。在溶劑中,由于靜電作用,鋰離子周圍會富集一定量的溶劑分子;溶劑分子的極性越大(即εr越大)富集量越多;而這些富集的溶劑分子將會在鋰離子之間附加額外的排斥作用使鋰離子更難相互靠近,因而使得r0,rmin增加而εmin減小;由于鋰離子周圍空間有限,只能容納有限的溶劑分子,溶劑化效應在εr較大的情況下將會趨于飽和,因而當εr較大時r0,rmin和εmin的變化較為平緩;依據(jù)圖4,當溶劑化效應趨于飽和時,鋰離子的溶劑化直徑為0.45~0.50 nm。
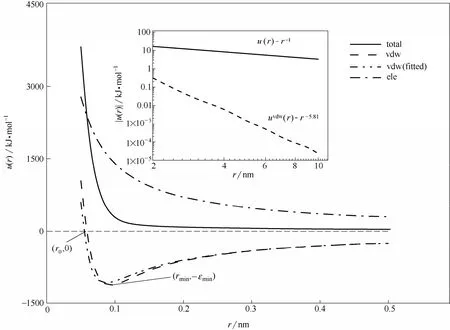
圖2 真空中鋰離子間相互作用勢能Fig. 2 Interacting potential between lithium ions in vacuum
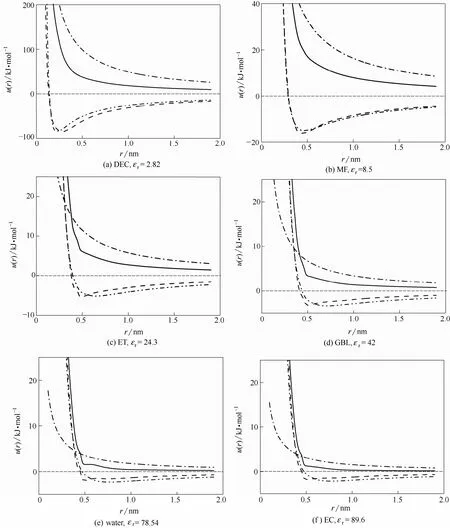
圖3 溶劑中鋰離子間相互作用Fig. 3 Interactions for lithium ions in solutions
在超級電容器充放電過程中,當電壓較低時(mV級),電極處鋰離子間距較大,此時鋰離子間相互作用由長程作用uele(r)主導;由式(14)及圖3可知,隨著介電常數(shù)的增加,長程排斥減弱,此時具有高介電常數(shù)的電解液更有利于鋰離子的吸附與儲存。而當電壓較高時(V級),電極處鋰離子間距較小,此時短程排斥起主導作用;由圖3和圖4可知,隨著介電常數(shù)的增加,短程排斥增強,此時具有低介電常數(shù)的電解液更有利于鋰離子的吸附與儲存。
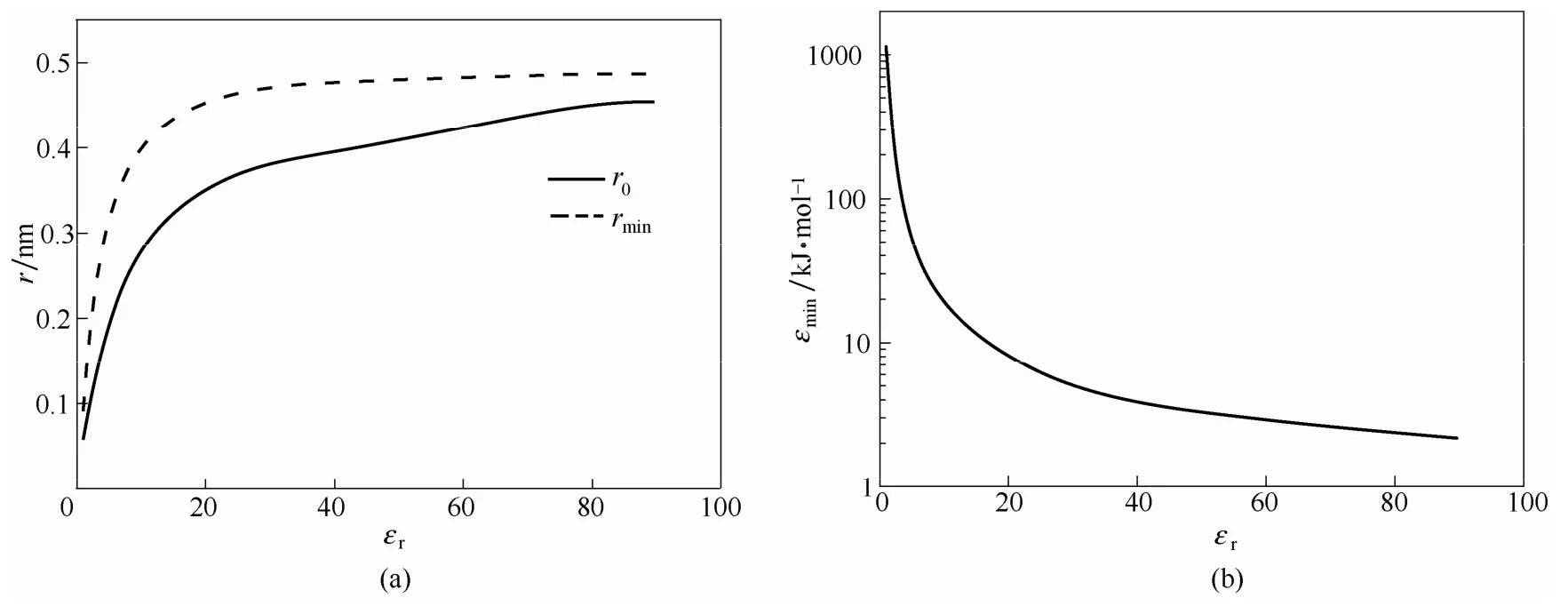
圖4r0、rmin以及εmin與相對介電常數(shù)的關系Fig. 4 Relationship betweenr0,rmin,εminand relative dielectric constant
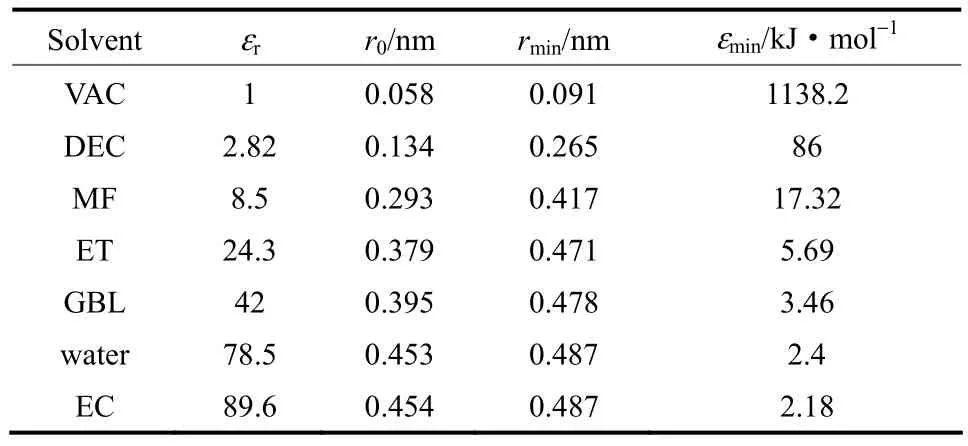
表1 不同溶劑中鋰離子間兩體作用零點能坐標r0、勢阱坐標rmin及勢阱深度εminTable 1 Coordinates of zero point energyr0, coordinates of potential wellrminand well depthεminof two-body interaction between lithium ions in different solvents
在隱式溶劑模型中,由于鋰離子的范德華相互作用勢受到溶劑化效應的顯著影響,因此有必要建立考慮溶劑化效應的鋰離子分子模型。為此,以
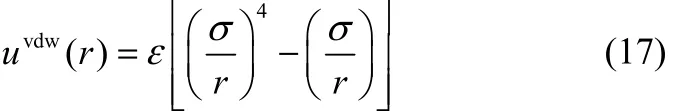
為擬合函數(shù),對以上6種溶劑中鋰離子相互作用勢以及真空中相互作用勢(短程區(qū)間)進行擬合,確定了ε、σ與εr的經(jīng)驗性函數(shù)關系式

如圖3所示,擬合后的勢能曲線與DFT計算得到的勢能曲線能很好地吻合,因此,可以期待基于式(17)和式(18)所構建的適用于不同溶劑條件下的鋰離子分子模型能很好地預測鋰離子電容器的特性。
2.2 鋰離子三體相互作用
如上所述,分子間相互作用除了經(jīng)典的對加和貢獻外還有量子多體貢獻。對于大多數(shù)系統(tǒng)而言,三體及三體以上的貢獻較小,可以忽略不計。但超級電容器和鋰電池中的鋰離子高度富集,離子間距離較小,量子效應可能比較明顯,為此考察了鋰離子三體相互作用。如圖5所示,考慮含有3個鋰離子的系統(tǒng),鋰離子以正三角形排列以模擬平面電極板附近鋰離子六方密堆積排列。

圖5 鋰離子三體作用模型Fig. 5 Three-body interaction model for lithiumions
根據(jù)式(2),三體作用勢u(3)(r)可通過從N= 3的體系總能量中扣除自能以及對加和能計算,即
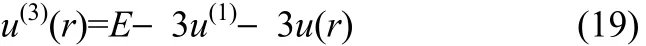
圖6所展示的即為真空中鋰離子三體相互作用勢。由圖可見,三體作用勢為吸引相互作用,作用范圍大約為r< 0.1 nm,強度約為兩體相互作用的1/3左右。這也說明了當鋰離子在電極附近高度富集時,三體相互作用可能對系統(tǒng)的熱力學和動力學性質造成一定的影響;而其作用效果為增加鋰離子間吸引相互作用。通過對三體作用勢進行雙對數(shù)擬合發(fā)現(xiàn),u(3)(r)與r的七次方呈反比,說明三體相互作用是短程相互作用,其衰減比色散作用更快。
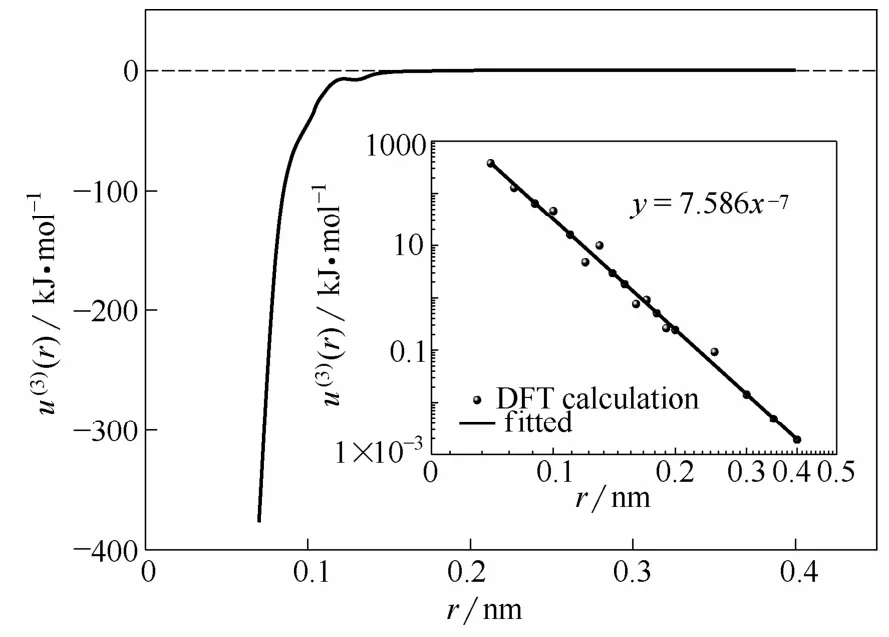
圖6 鋰離子三體相互作用勢能Fig. 6 Three-body interacting potential for lithium ions
3 結 論
本文采用量子密度泛函理論計算了超級電容器和鋰電池中鋰離子間的相互作用勢,考察了溶劑化效應對鋰離子間相互作用的影響,建立了適用于不同溶劑的鋰離子分子模型。計算結果表明,在短程區(qū)域內,鋰離子間的范德華相互作用能對靜電排斥作用產(chǎn)生強烈的屏蔽效應;而在長程區(qū)域內,靜電相互作用是鋰離子間相互作用的主導。在溶劑化條件下,隨著溶劑介電常數(shù)的增加,鋰離子間范德華作用勢阱迅速減小,而溶劑化直徑則呈現(xiàn)增大的趨勢。通過對計算結果進行擬合發(fā)現(xiàn),在短程區(qū)域內,鋰離子間范德華相互作用可以通過LJ-4-1勢能模型進行描述;而在長程區(qū)域內范德華作用的衰減速率與色散作用的衰減速率一致。另外,本文還考察了鋰離子間三體相互作用勢,發(fā)現(xiàn)三體相互作用為吸引相互作用;且僅當r< 0.1 nm時才對系統(tǒng)總能量產(chǎn)生顯著的影響;其衰減速率為七次方衰減,比色散作用衰減更快。
本文建立的鋰離子間相互作用勢模型可以進一步用于超級電容器和鋰電池電極性能的模擬或理論預測。
[1] FUTABA D N, HATA K, YAMADA T, et al.Shape-engineerable and highly densely packed single-walled carbon nanotubes and their application as super-capacitor electrodes[J]. Nature Materials, 2006, 5(12): 987-994.
[2] LV P, FENG Y Y, LI Y, et al.Carbon fabric-aligned carbon nanotube/MnO2/conducting polymers ternary composite electrodes with high utilization and mass loading of MnO2for super-capacitors[J]. Journal of Power Sources, 2012, 220: 160-168.
[3] NOKED M, OKASHY S, ZIMRIN T, et al.Composite carbon nanotube/carbon electrodes for electrical double-layer super capacitors[J]. Angewandte Chemie-International Edition, 2012, 51(7): 1568-1571.
[4] JIANG J, SHI W, SONG S, et al.Solvothermal synthesis and electrochemical performance in super-capacitors of Co3O4/C flower-like nanostructures[J]. Journal of Power Sources, 2014, 248: 1281-1289.
[5] KUMAR R, KIM H J, PARK S, et al.Graphene-wrapped and cobalt oxide-intercalated hybrid for extremely durable super-capacitor with ultrahigh energy and power densities[J]. Carbon, 2014, 79: 192-202.
[6] NISHIMOTO Y, YOKOGAWA D, YOSHIKAWA H, et al.Super-reduced polyoxometalates: excellent molecular cluster battery components and semipermeable molecular capacitors[J]. Journal of the American Chemical Society, 2014, 136(25): 9042-9052.
[7] TANG W, PENG L, YUAN C, et al.Facile synthesis of 3D reduced graphene oxide and its polyaniline composite for super capacitor application[J]. Synthetic Metals, 2015, 202: 140-146.
[8] ZOU J Y, ZHANG L, SONG J Y. Development of the 40 V hybrid super-capacitor unit[J]. IEEE Transactions on Magnetics, 2005, 41(1): 294-298.
[9] BORENSTIEN A, NOKED M, OKASHY S, et al.Composite carbon nano-tubes (CNT)/activated carbon electrodes for non-aqueous super capacitors using organic electrolyte solutions[J]. Journal of the Electrochemical Society, 2013, 160(8): A1282-A1285.
[10] DE D, KLUMPNER C, PATEL C, et al.Modelling and control of a multi-stage interleaved DC-DC converter with coupled inductors for super-capacitor energy storage system[J]. IET Power Electronics, 2013, 6(7): 1360-1375.
[11] WANG J D, PENG T J, SUN H J, et al.Effect of the hydrothermal reaction temperature on three-dimensional reduced graphene oxide's appearance, structure and super capacitor performance[J]. Acta Physico-Chimica Sinica, 2014, 30(11): 2077-2084.
[12] CAO X H, SHI Y M, SHI W H, et al.Preparation of MoS2-coated three-dimensional graphene networks for high-performance anode material in lithium-ion batteries[J]. Small, 2013, 9(20): 3433-3438.
[13] WANG H W, ZHANG Y, ANG H X, et al.A high-energy lithium-ion capacitor by integration of a 3D interconnected titanium carbide nanoparticle chain anode with a pyridine-derived porous nitrogen-doped carbon cathode[J]. Advanced Functional Materials, 2016, 26(18): 3082-3093.
[14] JIANG H, ZHANG H X, FU Y, et al.Self-volatilization approach to mesoporous carbon nanotube/silver nanoparticle hybrids: the role of silver in boosting Li ion storage[J]. ACS Nano, 2016, 10(1): 1648-1654.
[15] JENSEN K P, JORGENSEN W L. Halide, ammonium, and alkali metal ion parameters for modeling aqueous solutions[J]. Journal ofChemical Theory and Computation, 2006, 2(6): 1499-1509.
[16] GANCHEFF J S, KREMER C, VENTURA O N. Interaction of simple ions with water: theoretical models for the study of ion hydration[J]. Journal of Chemical Education, 2009, 86(12): 1403-1407.
[17] RIZZO R C, JORGENSEN W L. OPLS all-atom model for amines: resolution of the amine hydration problem[J]. Journal of the American Chemical Society, 1999, 121(20): 4827-4836.
[18] KAMINSKI G A, FRIESNER R A, TIRADO-RIVES J, et al.Evaluation and reparametrization of the OPLS-AA force field for proteinsviacomparison with accurate quantum chemical calculations on peptides[J]. Journal of Physical Chemistry B, 2001, 105(28): 6474-6487.
[19] ZHOU L, ZHAO D Y, LOU X W. Double-shelled CoMn2O4hollow microcubes as high-capacity anodes for lithium-ion batteries[J]. Advanced Materials, 2012, 24(6): 745-748.
[20] MADRIA N, ARUNKUMAR T A, NAIR N G, et al.Ionic liquid electrolytes for lithium batteries: synthesis, electrochemical, and cytotoxicity studies[J]. Journal of Power Sources, 2013, 234: 277-284.
[21] DEWAR M J S, ZOEBISCH E G, HEALY E F, et al.AM1: a new general purpose quantum mechanical molecular model[J]. Journal of the American Chemical Society, 1985, 107(13): 3902-3909.
[22] MURPHY R B, PHILIPP D M, FRIESNER R A. A mixed quantum mechanics/molecular mechanics (QM/MM) method for large-scale modeling of chemistry in protein environments[J]. Journal of Computational Chemistry, 2000, 21(16): 1442-1457.
[23] TAYLOR J, GUO H, WANG J.Ab initiomodeling of quantum transport properties of molecular electronic devices[J]. Physical Review B, 2001, 63(24): 245407.
[24] CHIBA M, FEDOROV D G, KITAURA K. Polarizable continuum model with the fragment molecular orbital-based time-dependent density functional theory[J]. Journal of Computational Chemistry, 2008, 29(16): 2667-2676.
[25] COHEN A J, MORI-SANCHEZ P, YANG W. Challenges for density functional theory[J]. Chemical Reviews, 2012, 112(1): 289-320.
[26] JOUNG I S, CHEATHAM T E Ⅲ. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations[J]. Journal of Physical Chemistry B, 2008, 112(30): 9020-9041.
[27] PEZESHKI S, LIN H. Molecular dynamics simulations of ion solvation by flexible-boundary QM/MM: on-the-fly partial charge transfer between QM and MM subsystems[J]. Journal of Computational Chemistry, 2014, 35(24): 1778-1788.
[28] HOHENBERG P, KOHN W. Inhomogeneous electron gas[J]. Physical Review B, 1964, 136(3B): B864-B871.
[29] KOHN W, SHAM L J. Self-consistent equations including exchange and correlation effects[J]. Physical Review, 1965, 140(4A): A1133-A1138.
[30] HO G S, LIGNERES V L, CARTER E A. Introducing profess: a new program for orbital-free density functional theory calculations[J]. Computer Physics Communications, 2008, 179(11): 839-854.
[31] FREEMAN F, HEHRE W J. Anab initiomolecular orbital theory and density functional theory study of the conformational free energies of methyltetrahydro-2H-thiopyrans[J]. Journal of Molecular Structure-Theochem, 2000, 529: 225-239.
[32] CARLING K M, CARTER E A. Orbital-free density functional theory calculations of the properties of Al, Mg and Al-Mg crystalline phases[J]. Modelling and Simulation in Materials Science and Engineering, 2003, 11(3): 339-348.
[33] LANGRETH D C, PERDEW J P. Exchange-correlation energy of a metallic surface: wave-vector analysis. ii[J]. Physical Review B, 1982, 26(6): 2810-2818.
[34] VOSKO S H, WILK L, NUSAIR M. Accurate spin-dependent electron liquid correlation energies for local spin-density calculations — a critical analysis[J]. Canadian Journal of Physics, 1980, 58(8): 1200-1211.
[35] PERDEW J P, WANG Y. Accurate and simple analytic representation of the electron-gas correlation-energy[J]. Physical Review B, 1992, 45(23): 13244-13249.
[36] ALONSO J A, GIRIFALCO L A. Nonlocal approximation to exchange potential and kinetic-energy of an inhomogeneous electron-gas[J]. Physical Review B, 1978, 17(10): 3735-3743.
[37] KERKER G P. Nonlocal-density approximation to exchange and correlation: effect on the silicon band structure[J]. Physical Review B (Condensed Matter), 1981, 24(6): 3468-3473.
[38] CUEVAS-SAAVEDRA R, CHAKRABORTY D, RABI S, et al.Symmetric nonlocal weighted density approximations from the exchange-correlation hole of the uniform electron gas[J]. Journal of Chemical Theory and Computation, 2012, 8(11): 4081-4093.
[39] PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple[J]. Physical Review Letters, 1996, 77(18): 3865-3868.
[40] BECKE A D. Density-functional thermochemistry(Ⅲ): The role of exact exchange[J]. The Journal of Chemical Physics, 1993, 98(7): 5648-5652.
[41] CHENGTEH L, WEITAO Y, PARR R G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density[J]. Physical Review B (Condensed Matter), 1988, 37(2): 785-789.
Construction of interaction model for lithium ion in super capacitors and lithium battery
LIU Yu1, JIANG Hao2, LI Chunzhong2, LIU Honglai3
(1State Key Laboratory of Chemical Engineering,School of Chemical Engineering,East China University of Science and Technology,Shanghai200237,China;2Key Laboratory for Ultrafine Materials of Ministry of Education,School of Materials Science and Engineering,East China University of Science and Technology,Shanghai200237,China;3State Key Laboratory of Chemical Engineering,School of Chemistry & Molecular Engineering,East China University of Science and Technology,Shanghai200237,China)
Lithium ions are highly concentrated in super capacitors and lithium battery, and to construct an accurate interaction model for lithium ions plays an important guiding role in predicting the performance of super capacitors and lithium battery and designing of electrode material. In this work, the interaction model was established for lithium ion in super capacitors and lithium battery by employing quantum density functional theory. It was focused on the characteristics and the solvation effect on the van der Waals interaction between lithium ions. It was revealed that the Coulombic repulsion was highly screened by the van der Waals interaction, and the solvation effect resulted a prodigious contribution to the van der Waals interaction. The molecular interaction model for lithium ion in different solvents was established by numerical fitting (primitive model). Besides, the three-body interaction for lithium ions was also considered and was revealed that the three-body interaction was an attraction, which was effective only when the ions were highly concentrated.
lithium ion;super capacitor;density functional theory;model;solvents;microscale
LIU Honglai, hlliu@ecust.edu.cn
O 642.4
:A
:0438—1157(2017)02—0552—08
10.11949/j.issn.0438-1157.20160901
2016-07-01收到初稿,2016-11-29收到修改稿。
聯(lián)系人: 劉洪來。
:劉宇(1984—),男,博士。
國家自然科學基金項目(91334203,21506051);上海市浦江人才計劃項目(15PJ1401400);中央高校基本科研業(yè)務費項目(222201414008);化學工程聯(lián)合國家重點實驗室開放課題項目(SKL-ChE-15C05)。
Received date: 2016-07-01.
Foundation item: supported by the National Natural Science Foundation of China (91334203, 21506051), the Shanghai Pujiang Program (15PJ1401400), the Fundamental Research Funds for the Central Universities of China (222201414008) and the Open Project of State Key Laboratory of Chemical Engineering (SKL-ChE-15C05).
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
網(wǎng)絡安全與數(shù)據(jù)管理(2022年1期)2022-08-29 03:15:20
導航定位學報(2022年4期)2022-08-15 08:27:00
中學生數(shù)理化·中考版(2022年8期)2022-06-14 06:55:24
新世紀智能(數(shù)學備考)(2021年9期)2021-11-24 01:14:36
成都醫(yī)學院學報(2021年2期)2021-07-19 08:35:14
新世紀智能(數(shù)學備考)(2020年9期)2021-01-04 00:25:14
中學生數(shù)理化·七年級數(shù)學人教版(2020年10期)2020-11-26 08:24:50
數(shù)學物理學報(2020年2期)2020-06-02 11:29:24
光學精密工程(2016年6期)2016-11-07 09:07:19
