市售保健品中7種醒酒護肝功效成分的毛細管電泳測定
2016-12-27 01:22:40s鄒海民孫成均李永新楊曉松曾紅燕
高等學校化學學報 2016年7期
關鍵詞:實驗
s鄒海民, 周 琛, 孫成均, 李永新, 楊曉松, 文 君, 曾紅燕
(1. 四川大學華西公共衛生學院, 成都 610041; 2. 成都市疾病預防控制中心, 成都 610047)
?
市售保健品中7種醒酒護肝功效成分的毛細管電泳測定
s鄒海民1,2, 周 琛1, 孫成均1, 李永新1, 楊曉松2, 文 君2, 曾紅燕1
(1. 四川大學華西公共衛生學院, 成都 610041; 2. 成都市疾病預防控制中心, 成都 610047)
采用膠束電動毛細管電泳-二極管陣列檢測器同時檢測了市售保健品中兒茶素、 表兒茶素、 表沒食子兒茶素、 表兒茶素沒食子酸酯、 表沒食子兒茶素沒食子酸酯、 二氫楊梅素和甘草酸等7種醒酒護肝功效成分. 采用正交設計法對毛細管電泳條件[緩沖劑濃度、 添加劑十二烷基硫酸鈉(SDS)濃度、 添加劑乙腈比例以及電泳緩沖溶液pH]進行了優化. 在優化的條件下, 7種組分在各自的線性范圍內相關系數r≥0.9989, 檢出限和定量限分別為0.26~2.22 μg/g(S/N=3)和0.87~7.39 μg/g(S/N=10), 日內精密度和日間精密度分別為1.3%~2.5%和1.9%~3.9%, 樣品加標回收率為91.4%~104.9%, 加標樣品的相對標準偏差(RSD)在1.4%~3.2%之間. 本方法可在8 min內實現7種功效成分的同時檢測, 能夠滿足市售醒酒護肝產品的常規分析和質量評價要求.
膠束電動毛細管電泳; 正交設計; 兒茶素; 二氫楊梅素; 甘草酸
目前, 市售醒酒護肝功能食品多為各種茶葉或其提取物, 其主要成分包括兒茶素類[1]、 二氫楊梅素[2]和甘草酸[3,4]等. 兒茶素類主要包括兒茶素(C)、 表兒茶素(EC)、 表沒食子兒茶素(EGC)、 表兒茶素沒食子酸酯(ECG)和表沒食子兒茶素沒食子酸酯(EGCG)等. 兒茶素類化合物的化學結構中均含有較多的酚羥基[5], 能與體內乙醇代謝過程中產生的自由基結合而將其清除, 具有較好的解酒效果[6]; 該類化合物也有較強的抗氧化能力, 能有效緩解酒精造成的肝脂質過氧化[7]. 二氫楊梅素(DMY)又名蛇葡萄素, 為天然多酚羥基雙氫黃酮醇類化合物, 大量存在于葡萄屬植物藤茶中. 歐賢紅等[8]通過體外實驗證實二氫楊梅素能有效清除氧陰離子自由基和羥自由基, 具有較強的抗氧化活性. 甘草酸(GA)是中草藥甘草的主要活性成分, 屬三萜類化合物. 甘草不僅有抑制肝臟脂質過氧化、 清除自由基和抗肝纖維化的功效[9], 而且在中國傳統復方上常作為一味獨特的導藥, 可協同加強其它組分的功效[10]. 兒茶素類、 二氫楊梅素和甘草酸的常見分析方法有薄層色譜法[11]、 氣相色譜法[12]、 高效液相色譜法[13,14]和毛細管電泳法[15,16]等, 其中毛細管電泳分離模式多、 分析速度快、 分離效率高、 消耗樣品少且能滿足快速分析的需要. 目前文獻[1,4,6,15]多只針對某一種或一類醒酒護肝成分進行檢測, 而對于黃烷醇類、 黃酮醇類及三萜類等多種成分同時檢測的報道較少. 本文采用毛細管電泳法同時測定市售保健品中多種醒酒護肝功效成分. 與傳統的高效液相色譜法相比, 本方法具有樣品用量少、 分析時間短、 特異性好、 環境污染小、 靈敏度高及重現性好等優點, 可用于批量樣品的檢測, 能滿足市售保健品中多種醒酒護肝功效成分的常規分析和質量評價.
1 實驗部分
1.1 試劑與儀器
兒茶素、 表兒茶素、 表沒食子兒茶素、 表兒茶素沒食子酸酯、 表沒食子兒茶素沒食子酸酯、 二氫楊梅素和甘草酸(純度≥98%, 成都曼思特生物科技有限公司); 四硼酸鈉(Na2B4O7, 分析純, 成都化學試劑廠); 十二烷基硫酸鈉(SDS, 分析純, 天津大茂化學試劑廠); 甲醇(色譜純, 天津市科密歐化學試劑有限公司); 乙腈(ACN, 高效液相色譜級, Sigma-Aldrich公司).
P/ACE MDQ型高效毛細管電泳儀, 配有光電二極管陣列檢測器(美國Beckman公司); pHS-2C型數字酸度計(成都世紀方舟科技有限公司); BSA224S型電子天平(0.1 mg, 賽多利斯科學儀器有限公司); 754系列紫外-可見分光光度計(美國Labtech公司); KQ-250型超聲波清洗器(昆山市淀山湖儀器檢測廠).
1.2 溶液配制
將7種標準物質均以甲醇為溶劑配制成1 mg/mL的標準儲備液.
標準溶液的配制: 分別吸取一定體積的各標準儲備液, 用毛細管電泳緩沖液定容至10 mL, 配制成含GA為400 μg/mL, C, EC, ECG, EGC, EGCG和DMY均為100 μg/mL的混合標準儲備液, 使用前用電泳緩沖液進一步稀釋成標準系列. 所有標準溶液均需在4 ℃下避光保存, 至少可以穩定30 d.
1.3 實驗方法
1.3.1 毛細管電泳實驗 毛細管電泳條件: 熔融石英毛細管(50 cm×50 μm, 有效長度為40 cm, 未涂層, Beckman公司); 電泳緩沖液由25 mmol/L Na2B4O7, 35 mmol/L SDS和7%(體積分數)ACN組成, 其pH=7.00, 經0.45 μm微孔濾膜過濾、 超聲波脫氣10 min后備用; 分離電壓為25 kV, 采用光電二極管陣列(PDA)檢測器在190~400 nm范圍內全掃描檢測. GA選擇254 nm處色譜峰進行定量分析, 其余組分均選擇210 nm; 分析溫度為25 ℃; 3.45 kPa壓力下進樣5 s.
為保證實驗結果的重現性, 實驗前依次用0.1 mol/L NaOH溶液、 超純水和緩沖溶液沖洗毛細管柱3 min; 每次分析結束后用電泳緩沖液沖洗毛細管柱3 min后進行下一次分析; 每進行3次分析后需更換兩極電泳緩沖液.
1.3.2 樣品前處理 將茶葉、 膠囊等固體樣品內容物取出后碾磨成粉, 過100目篩后, 稱取1 g, 用25 mL 80%(體積分數)的甲醇溶液超聲提取30 min; 直接吸取1.00 mL液態樣品用甲醇溶液定容至5.00 mL. 所有樣品溶液均以10000 r/min轉速離心10 min, 取上層清液設定的電泳條件進樣分析.
2 結果與討論
2.1 毛細管電泳條件的優化
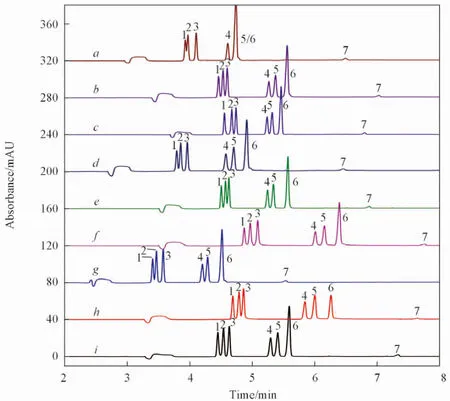
Fig.1 Electrophoretograms of 7 components in mixed standard solution with orthogonal experiment of L9(34)
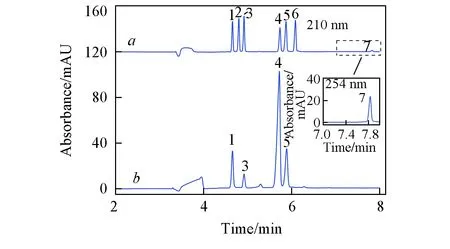
Fig.2 Electrophoretograms of mixed standard solution(a) and sample 2#(b)
影響毛細管電泳分離效果的因素較多, 正交設計法能同時考慮多個因素的交互作用, 具有簡便、 直觀且快速等優點. 本文在單因素預實驗的基礎上, 采用正交設計法進一步優化以期獲得最佳分離條件. 選取對各組分分離度影響較大的4個因素: 緩沖液硼砂濃度(20, 25和30 mmol/L)、 添加劑SDS濃度(25, 30和35 mmol/L)、 添加劑乙腈比例(5%, 7%和9%, 體積分數)以及電泳緩沖液pH(6.80, 7.00, 7.20), 進行L9(34)正交實驗設計, 各因素組合條件實驗見表S1(見本文支持信息).
各次條件實驗結果如圖1所示, 由于7種組分的出峰時間均較短能滿足分析要求, 因此本文僅以難分離對EGC/C, C/EC, EGCG/ECG和ECG/DMY的分離度(Rs)作為評定條件實驗效能的標準, 采用SPSS 17.0軟件進行統計分析(結果見表S1). 每個水平i(i=1, 2, 3)的平均分離度值Ki和極差R的計算公式如下:
Ki=$∑$R\%s\%i/3,R=Kmax-Kmin
式中,Kmax代表Ki的最大值;Kmin代表Ki的最小值.
Kmax代表的i水平是能獲得最大分離度的條件, 不同分離因素所計算出的R值不同,R值越大則表示該因素對難分離組分的分離度影響越大, 反之亦然.
由圖1可見, 在9次實驗中, 7種組分出峰時間均小于8 min, 其中第2, 3, 6和9次實驗各組分均達到基線分離. 由表S1可知, 影響EGC/C分離度的主要因素為緩沖液的pH, 當pH=7.00時分離度最好; 影響C/EC分離度的主要因素也為緩沖液的pH, 當pH=6.80時分離度最好; 影響EGCG/ECG分離度的主要因素為SDS濃度, 當SDS濃度為30 mmol/L時分離度最好; 而影響ECG/DMY分離度的主要是乙腈比例, 當乙腈體積分數為5%時分離度最好. 由表S1還可知, EGC/C和C/EC這2對組分的分離度較小, 而EGCG/ECG和ECG/DMY這2對組分的分離度相對更好, 因此優先考慮各因素對分離度較小的EGC/C和C/EC的影響, 以各組分均能達到基線分離(分離度≥1.50)為前提對各因素進行優選, 通過SPSS 17.0軟件分析, 最終確定優化的實驗條件為A2B3C2D2, 即緩沖液由25 mmol/L硼砂、 35 mmol/L SDS和7%乙腈組成, pH=7.00. 在優化條件下對7種目標組分進行電泳分析, 結果如圖2所示, 可見各組分均達到基線分離.
2.2 定量分析波長的選擇
對目標分析物于設定的電泳條件下分別單標進樣分析, 用PAD檢測器在190~400 nm波長范圍內全掃描. 根據各化合物的紫外吸收光譜得到兒茶素類5種化合物的最大吸收波長均為210 nm, 次吸收波長為280 nm, 由于EGC在280 nm處紫外吸收非常低, 故兒茶素類5種物質選擇210 nm為定量分析波長; 二氫楊梅素的最大吸收波長為210 nm, 次吸收波長290 nm, 選擇210 nm為其定量分析波長; 選擇甘草酸的最佳吸收波長254 nm為其定量分析波長.
2.3 樣品超聲提取條件優化
超聲波輔助提取法是利用超聲波具有的機械效應、 空化效應及熱效應來強化提取效果的方法[17]. 該方法具有提取效率高、 提取時間短、 操作簡單和適應性廣等優點, 可有效避免高溫高壓對待測組分的破壞, 在天然產物有效成分的提取中應用較為廣泛[18]. 影響超聲提取效能的因素有提取劑種類、 提取時間、 提取溫度、 樣品顆粒粒徑和固液比等. 由于待測組分中的兒茶素類和二氫楊梅素對熱不穩定[19,20], 所以實驗選擇常溫下進行提取. 在預實驗的基礎上選取對提取效率影響較大的因素----提取劑甲醇濃度(A, 70%, 80%和90%, 體積分數), 超聲時間(B, 10, 20和30 min), 固液比(C, g/mL: 1∶15, 1∶20和1∶25)以及提取次數(D, 1, 2和3), 采用L9(34)正交設計法對主要因素進行優化.
將1#~4#固態樣品粉碎, 過100目篩, 混勻后備用. 取混合樣27份, 各約1.0 g, 按正交設計方案分別進行處理, 每種組合測定3個平行樣, 計算7種組分總含量的平均值, 采用SPSS 17.0軟件進行統計分析. 結果(表S2, 見本文支持信息)表明, 各因素對提取效率的影響程度順序為甲醇濃度>提取時間>固液比>提取次數, 其中提取劑甲醇濃度和提取時間為主要影響因素, 固液比和提取次數對提取效率無明顯影響. 當提取時間從10 min提高到20 min時, 提取效率有較大提高; 而20 min到30 min提取效率改變較小. 為縮短實驗周期, 選擇超聲提取時間為30 min, 不再增加提取時間; 同時由于提取次數對提取效率影響很小, 為減化提取過程, 實驗選擇只進行一次超聲提取. 因此, 最終選擇各因素最佳組合為A2B3C3D1, 即采用25 mL 80%的甲醇作為提取劑進行一次提取, 提取時間為30 min. 在最終優化的提取條件下對相同樣品進行提取, 各組分總的含量為2.237 mg/g.
2.4 毛細管電泳分離
在優化的毛細管電泳分離條件和超聲提取條件下, 標準溶液及樣品的電泳譜圖見圖2. 標準中各組分均達到基線分離且出峰時間均在8 min內, 樣品的測定采樣電泳遷移時間和光譜匹配度進行定性, 峰面積進行定量.
2.5 方法學指標
2.5.1 回歸方程、 線性范圍和檢出限 取系列混合標準溶液分別進樣分析, 每個濃度重復測定3次, 以待測物濃度(x, μg/mL)為橫坐標, 峰面積平均值(y, mAU5min)為縱坐標繪制標準曲線. 測定結果見表1, 各標準曲線的相關系數均在0.9989~0.9995之間. 以3倍和10倍基線噪聲所對應的各組分含量計算出方法檢出限(LOD)和定量限(LOQ)分別為0.26~2.22 μg/g和0.87~7.39 μg/g.
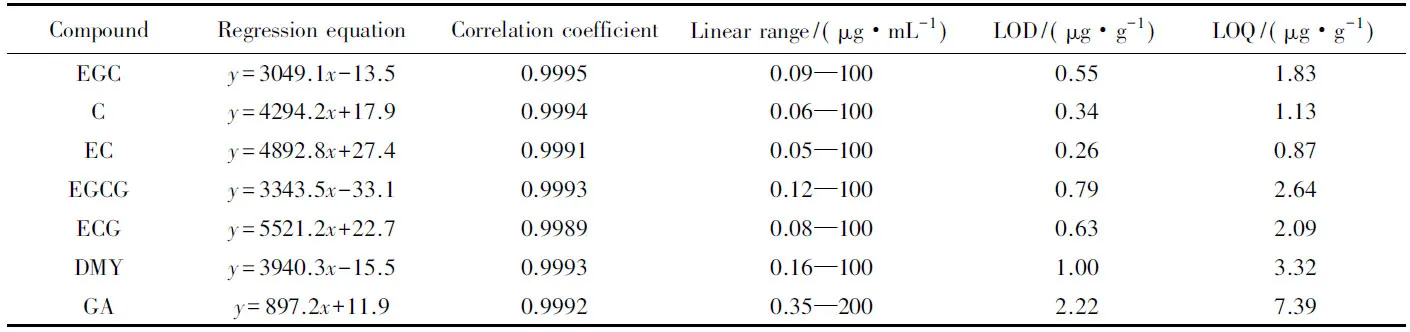
Table 1 Regression equation, linear range, LOD and LOQ of 7 components
2.5.2 精密度 取7種組分的混合標準溶液(C, EC, ECG, EGC, EGCG和DMY濃度為10 μg/mL, GA濃度為40 μg/mL)進行測定, 在同一天內連續進樣6次, 以峰面積和保留時間的相對標準偏差(RSD)考察日內精密度; 對同一混合標準溶液連續6 d進樣測定, 考察日間精密度. 結果(表2)顯示, 日內精密度和日間精密度分別為1.3%~2.5%和1.9%~3.9%, 滿足分析方法對精密度的要求.
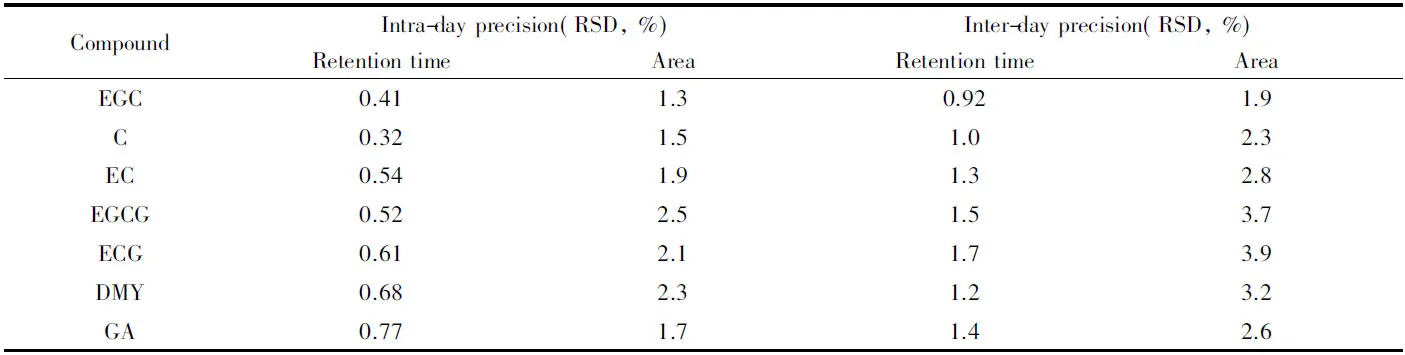
Table 2 Intra-day precision and inter-day precision(n=6)
2.5.3 加標回收率 將同一樣品分別加入3個不同濃度水平的標準溶液中, 每個濃度水平測定6個平行樣, 結果見表3. 回收率在91.4%~104.9%之間, 加標樣品的RSD在1.4%~3.2%之間, 表明該方法能滿足實際分析測定準確度的要求.
2.6 樣品檢測
稱取1 g固態樣品(精確至0.1 mg), 吸取1.00 mL液態樣品并稱重, 每份樣品測定3個平行樣,
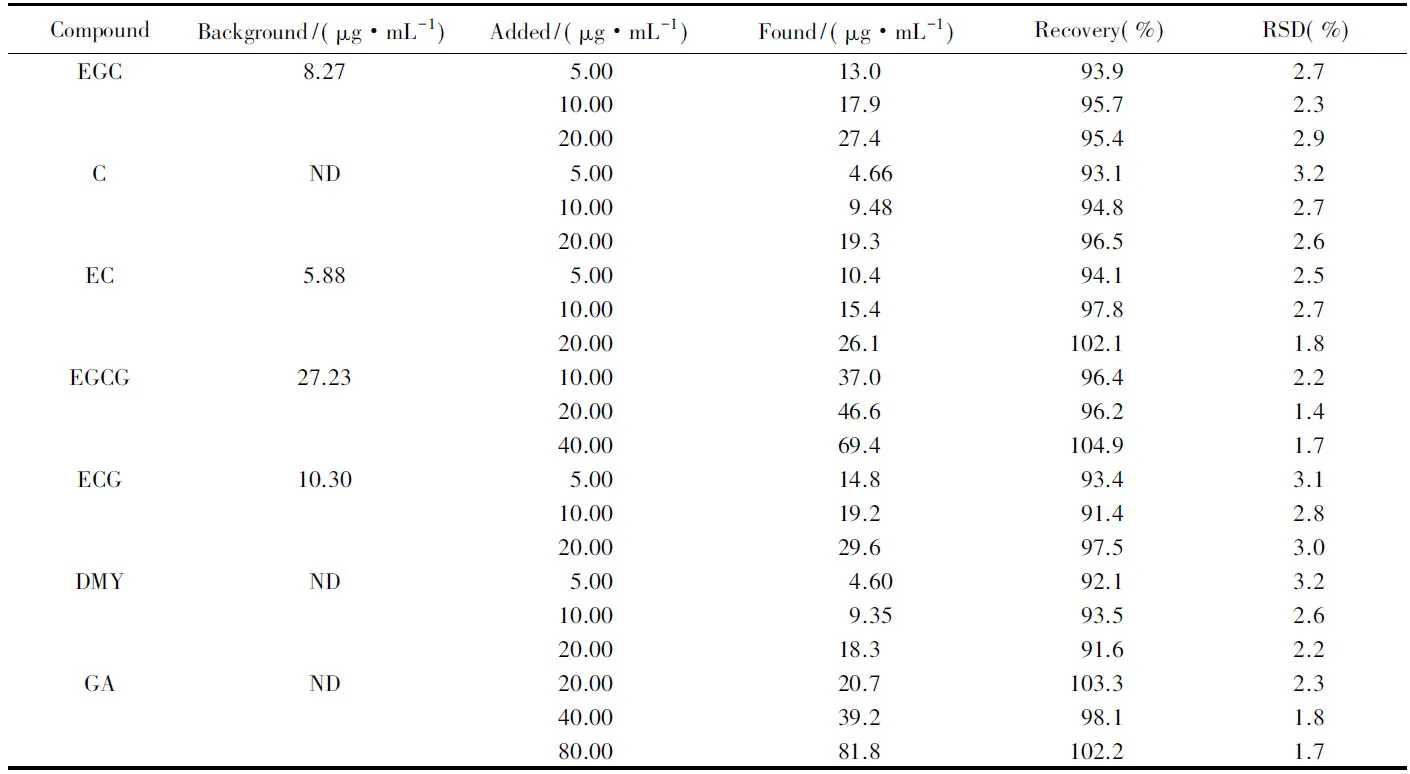
Table 3 Results of the spiked recovery tests*(n=6)
對樣品進行前處理后進樣, 測得樣品中各待測物的含量列于表4. 結果表明, 樣品中均含有多種功效成分. 所檢出的功效成分中兒茶素類的檢出頻率較高, 這可能與其在植物中廣泛存在有關. 雖然藤茶作為醒酒護肝產品應用的時間較短, 但二氫楊梅素在藤茶中的含量較高(表4, Sample 1), 研究[21]表明二氫楊梅素能有效清除自由基, 具有較強的抗氧化活性, 是護肝醒酒的良品, 說明藤茶可能具有較好的醒酒護肝功效, 具有一定的應用前景.
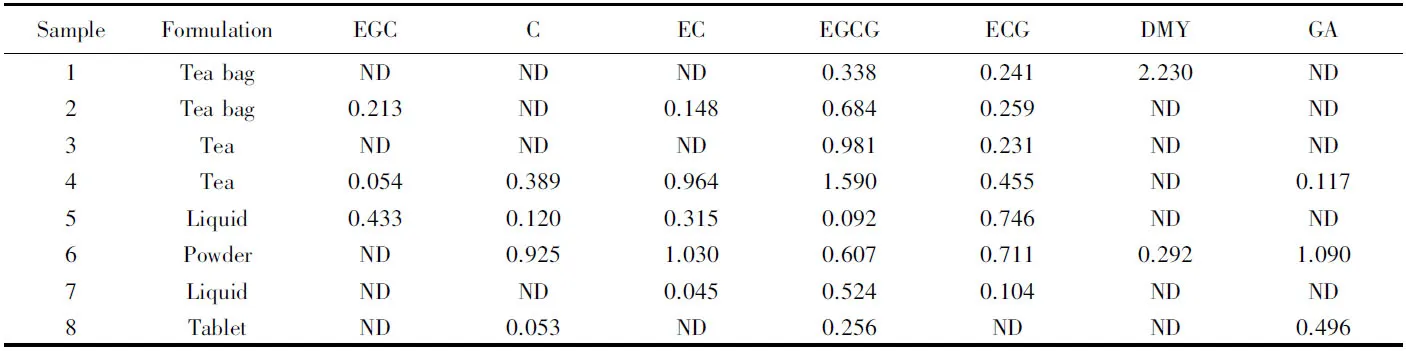
Table 4 Contents of the target components in 8 samples*(mg/g, n=3)
綜上所述, 建立了用毛細管電泳同時測定醒酒護肝產品中多種功效成分的方法, 并通過正交設計對實驗條件進行了優化, 該方法能在8 min內實現7種功效成分的基線分離. 與傳統的高效液相色譜法相比, 本文方法具有樣品需用量少、 分析時間短、 特異性好、 環境污染小、 靈敏度高且重現性好等優點, 可用于批量樣品的檢測, 能滿足市售保健品中多種醒酒護肝功效成分的常規分析和質量評價.
支持信息見http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20150980.
[2] Ye L. Y., Wang H. J., Duncan S. E., Eigel W. N., O’Keefe S. F.,FoodChem., 2015, 172, 416—422
[3] Charpe T. W., Rathod V. K.,FoodBioprod.Process, 2015, 93, 51—57
[4] Shabkhiz M. A., Eikani M. H., Bashiri Sadr Z., Golmohammad F.,FoodChem., 2016, 210, 396—401
[5] Liu G. Q., Dong J., Wang H., Wan L. R., Duan Y.S., Chen S. Z.,Chem.J.ChineseUniversities, 2009, 30(8), 1566—1570(劉國強, 董靜, 王弘, 萬樂人, 端裕樹, 陳世忠. 高等學校化學學報, 2009, 30(8), 1566—1570)
[6] Anissi J., El Hassouni M., Ouardaoui A., Sendide K.,FoodChem., 2014, 150, 438—447
[7] Oh J., Jo H., Cho A. R., Kim S. J., Hanet J.,FoodControl, 2013, 31(2), 403—409
[8] Ou X. H., Ye Y., Huang Q. J., Liu H. G., Song Y. F.,Nat.Prod.Res.Dev., 2013, 25(2), 245—248(歐賢紅, 葉勇, 黃秋潔, 劉華鋼, 宋云飛. 天然產物研究與開發, 2013, 25(2), 245—248)
[9] Fu Y., Chen J., Li Y. J., Zheng Y. F., Li P.,FoodChem., 2013, 141(2), 1063—1071
[10] Wang X.Y., Zhang H, Chen L. L., Shan L. H., Fan G. W., Gao X. M.,J.Ethnopharmacol., 2013, 150(3), 781— 790
[11] Boudesocque L., Dorat J., Pothier J., Gueiffer A., Gueiffier C. E.,FoodChem., 2013, 139, 866—871
[12] Soleas G. J., Yan J., Goldberg D. M.,J.Chromatogr.B, 2001, 757(1), 161—172
[13] Liao W. C., Lin Y. H., Chang T. M., Huang W. Y.,FoodChem., 2012, 132(4), 2188—2193
[14] Rahim A. A., Nofrizal S., Saad B.,FoodChem., 2014, 147, 262—268
[15] Peres R. G., Tonin F. G., Tavares M. F. M., Amaya D. B. R.,FoodChem., 2011, 127(2), 651—655
[16] Zhang S., Dong S. Q., Chi L. Z., He P. G., Wang Q. J., Fang Y. Z.,Talanta, 2008, 76(4), 780—784
[17] Vinatoru M.,Ultrason.Sonochem., 2001, 8(3), 303—313
[18] Morelli L. L. L., Prado M. A.,Ultrason.Sonochem., 2012, 19(6), 1144—1149
[19] Lun S. Y., Leung L. K., Huang Y., Chen Z. Y.,FoodChem., 2003, 83(2), 189—195
[20] Ananingsih V. K., Sharma A., Zhou W. B.,FoodRes.Int., 2013, 50(2), 469—479
[21] Xu J. J., Yao M. J., Wu M. C.,FoodSci., 2008, 29(11), 622—625(徐靜娟, 姚茂君, 鄔敏辰. 食品科學, 2008, 29(11), 622—625)
(Ed.: D, K)
Simultaneous Determination of 7 Components in Functional Food for Anti-hangover and Hepatoprotection by Capillary Electrophoresis?
? Supported by the 12th Five-year National Key Technology R & D Program, Ministry of Science and Technology of China(No.2012BAD33B02).
ZOU Haimin1,2, ZHOU Chen1, SUN Chengjun1, LI Yongxin1,YANG Xiaosong2, WEN Jun2, ZENG Hongyan1*
(1.WestChinaSchoolofPublicHealth,SichuanUniversity,Chengdu610041,China;2.ChengduCenterforDiseaseControl&Prevention,Chengdu610047,China)
A rapid method of micellar electrokinetic capillary electrophoresis(CE) coupled with diode array detection was developed for simultaneous determination of catechin, epicatechin, epigallocatechin, epicatechin gallate, epigallocatechin gallate, dihydromyricetin and glycyrrhizic acid in functional food for anti-hangover and hepatoprotection. The parameters of capillary electrophoresis, including buffer concentration, concentration of sodium dodecyl sulfate(SDS), volume ratio of acetonitrile, and pH of the buffer, were optimized with orthogonal design. Under the optimal analytical conditions, the peak area of each analyte and its concentration had a good correlation within the linear range(r≥0.9989). Limit of detection(LOD) and quantification(LOQ) of the method were in the range of 0.26—2.22 μg/g(S/N=3) and 0.87—7.39 μg/g(S/N=10), respectively. The intra- and inter-day relative standard deviations(RSDs) of the mixed standard solution were 1.3%—2.5% and 1.9%—3.9%, respectively. While the spiked recoveries of the analytes were 91.4%—104.9% and the RSDs of the spiked samples were 1.4%—3.2%. The method of capillary electrophoresis for determination of the 7 components in functional food for anti-hangover and hepatoprotection was proposed in this study and could achieve baseline separation for all the target components within 8 min. The results show that the method could meet the requirement for routine analysis and quality control and evaluation.
Micellar electrokinetic capillary electrophoresis; Orthogonal design; Catechin; Dihydromyricetin; Glycyrrhizic acid
2015-12-23.
日期: 2016-06-02.
科技部“十二五”科技支撐計劃(批準號: 2012BAD33B02)資助.
10.7503/cjcu20150980
O657.8
A
聯系人簡介: 曾紅燕, 女, 博士, 副教授, 主要從事理化檢驗方法研究. E-mail: zhm504532@163.com
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
