氣相色譜檢測水產品中有機氯類農藥殘留*1
2016-04-25 02:39:26杭學宇馮曉青
國際檢驗醫學雜志 2016年6期
關鍵詞:氣相色譜
宋 鑫,杭學宇,王 芹,王 露,馮曉青,徐 瑞,茅 力
(1.南京醫科大學公共衛生學院,江蘇南京 211166;2.淮安市疾病預防控制中心,江蘇淮安 223001)
?
氣相色譜檢測水產品中有機氯類農藥殘留*1
宋鑫1,2,杭學宇2,王芹1,2,王露2,馮曉青2,徐瑞2,茅力1△
(1.南京醫科大學公共衛生學院,江蘇南京 211166;2.淮安市疾病預防控制中心,江蘇淮安 223001)
摘要:目的建立氣相色譜定量測定水產品中16種有機氯類混合農藥殘留的方法。方法樣品由乙腈提取,以氨基固相萃取柱(Carb/NH2)凈化,經氣相色譜檢測分析。結果16種化合物在0.05~1.00 mg/L內線性關系良好(r>0.998),各有機氯的檢出限為0.04~0.31 μg/kg(S/N=3)。在加標水平為50、100、200 μg/kg時,該方法的回收率為72.6%~115.2%,相對標準偏差為0.6%~7.5%。結論該研究建立的方法選擇性好、靈敏度高,適用于水產品中有機氯類農藥的多殘留痕跡測定。
關鍵詞:氣相色譜;水產品;有機氯
近年來,水體污染越來越嚴重,水產品也不斷受到嚴重威脅,重金屬和有機物是其主要的污染物。因此,有必要對水產品進行污染物監測。有機氯農藥作為殺蟲劑,具有殺蟲效率高、易分解等特點,在提高糧食和蔬菜的產量與質量等方面起著積極的作用,為目前農業生產上控制病蟲害的主要農藥之一。但是,在使用過程中存在著許多問題,如超劑量使用,使用禁用產品,不按操作規程使用等。因此,很容易污染水體,從而造成水產品中有機氯殘留超標的問題,且有機氯可在人體內蓄積導致中毒,因此對有機氯農藥的監管尤為重要。目前,農藥殘留檢測方法主要有氣相色譜法[1-8]、氣相色譜-質譜法[9-11]、液相色譜-質譜法[12]等。但由于氣質聯類儀器用較為昂貴,因此氣相色譜儀還是檢測農藥殘留的主要儀器。本文建立了采用固相萃取凈化氣相色譜同時檢測水產品中16種有機氯農藥殘留的方法,現報道如下。
1材料與方法
1.1儀器與試劑(1)儀器:Agilent 6890N氣相色譜儀(美國Agilent公司);FSH-Ⅱ型高速電動勻漿器(環宇儀器公司);TGL-16 臺式高速冷凍離心機(湖南湘儀);Vortex-genie22T 渦旋振蕩器(上海凌初公司);Techne 氮吹儀(美國TECHNE公司);AR2130 電子天平(感量0.000 1 g);WSZ-20A振蕩器(上海-恒科技有限公司);DIKMA 氨基固相萃取柱(Carb/NH2,迪馬);LABOROTA4000eco 旋轉蒸發儀(德國)。(2)試劑:16種有機氯農藥單標(100 μg/mL,1 mL)均購自農業部環境保護科研監測所;乙腈、丙酮、正己烷(色譜純,默克);氯化鈉(分析純);無水硫酸鈉(分析純,使用前在650 ℃灼燒4 h,貯于干燥容器中,冷卻后備用)。16種有機氯農藥混合標準溶液的配制,精確吸取0.1 mL各有機氯農藥單標,用丙酮溶液定容至10 mL容量瓶中,配成1.0 mg/L混合標準使用液。精確吸取適量的混合標準使用液,用丙酮溶劑稀釋,配制成含16種有機氯農藥的質量濃度分別為0.05、0.10、0.20、0.50、1.00 mg/L的系列標準工作液。(3)受檢試樣:試樣采購自江蘇淮安地區市售的鯽魚、青蝦、螃蟹、河蚌等。分別取可食部分,勻漿后于-20 ℃下貯存備用。
1.2試驗方法
1.2.1樣品提取稱取勻漿樣品5.0 g于50 mL離心管中,加入5.0 g氯化鈉和20 mL乙腈,渦旋混勻1 min,超聲提取30 min,加入8.0 g無水硫酸鈉,渦旋混勻1 min,5 000 r/min離心5 min,取上清液于雞心瓶中。用10 mL乙腈重復提取一次,合并提取液,30 ℃水浴中減壓濃縮至近干,待凈化。
1.2.2固相萃取凈化用5 mL丙酮-正己烷(1∶9,v/v),5 mL正己烷依次活化氨基固相萃取柱(Carb/NH2),提取氣相色譜液用正己烷復融至5 mL后上樣并收集,用20 mL丙酮-正己烷(1∶9,v/v)洗脫,收集合并洗脫液,30 ℃水浴中減壓濃縮至近干,用正己烷定容至1 mL過膜,待氣相色譜分析測定。
1.2.3氣相色譜分析條件色譜柱為HP-5(30 m×0.32 mm,25 μm) (美國Agilent公司);電子捕獲檢測器(ECD),300 ℃。載氣為氮氣(純度大于99.999%);載氣流速為1.0 mL/min,尾吹50 mL/min。不分流進樣;進樣量為1 μL;柱溫程序采用程序升溫法,初始溫度為60 ℃,保持1 min;以40 ℃/min升至170 ℃;以2 ℃/min升至235 ℃;以40 ℃/min升至280 ℃。
1.3統計學處理采用SPSS19.0軟件進行數據處理及統計學分析。
2結果
2.1提取溶劑的選擇本試驗對乙腈、丙酮-二氯甲烷(1∶1,v/v)、丙酮-正己烷(1∶1,v/v)等提取溶劑對有機氯農藥的提取效率進行了考察。以河蚌肉進行加標回收試驗(加標水平為50 μg/kg,平行測定3次)進行對比,見圖1(見《國際檢驗醫學雜志》網站主頁“論文附件”)。從中可以看出不同的提取溶劑對大多數的有機氯農藥組分的提取效果相差不大。在對艾氏劑、b-六六六、狄氏劑等6種組分比較中,乙腈提取回收率為79.9%~95.3%,優于丙酮-正己烷(65.3%~85.3%)和丙酮-二氯甲烷(48.6%~72.3%),選用乙腈作為提取試劑。
2.2固相萃取柱選擇本試驗通過了對Carb/NH2、Florisil凈化柱及C18固相萃取柱進行加標試驗比較,以鯽魚肉進行加標回收試驗(加標水平為100 μg/kg,平行測定3次)進行對比。從中得出各組分回收率在使用Carb/NH2時為92.6%~100.5%,Florisil凈化柱為76.9%~89.3%,C18固相萃取柱為69.9%~81.3%,使用Carb/NH2凈化最優。
2.3氣相色譜測定16種有機氯農藥混合標準溶液(1.0 mg/L)和蟹肉基質加標(0.1 mg/L)的色譜圖見圖2、3。
2.4標準曲線和檢出限對混合標準溶液用丙酮稀釋,將16種有機氯農藥配制成質量濃度分別為0.05、0.10、0.20、0.50、1.00 mg/L的系列標準工作液進行氣相色譜測定。每個濃度點平行測定3次,以峰面積平均值對質量濃度進行線性回歸,得到的回歸方程見表1。各有機氯農藥的線性范圍在0.05~1.00 mg/L,相關系數均大于0.998。
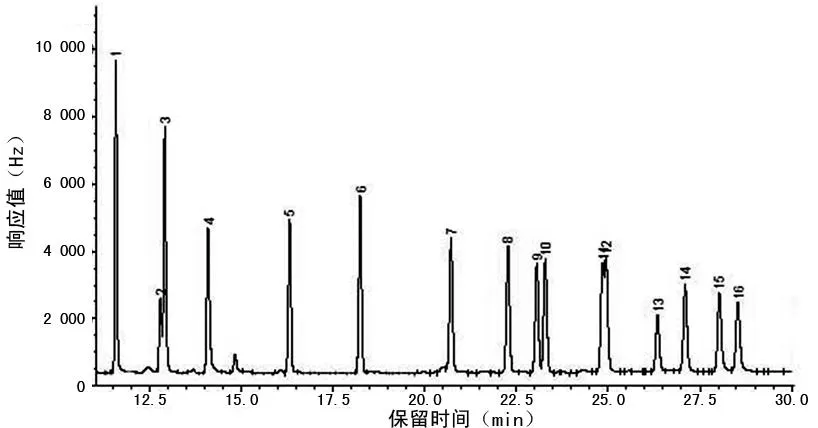
1:α-六六六;2:β-六六六;3:γ-六六六;4:δ-六六六;5:七氯;6:艾氏劑;7:環氧七氯;8:g-氯丹;9:硫丹Ⅰ;10:a-氯丹;11:p,p′-滴滴伊;12:狄氏劑;13:異狄劑;14:硫丹Ⅱ;15:p,p′-滴滴滴;16:p,p′-滴滴涕。
圖216種有機氯農藥混合標準溶液(1.0 mg/L)色譜圖
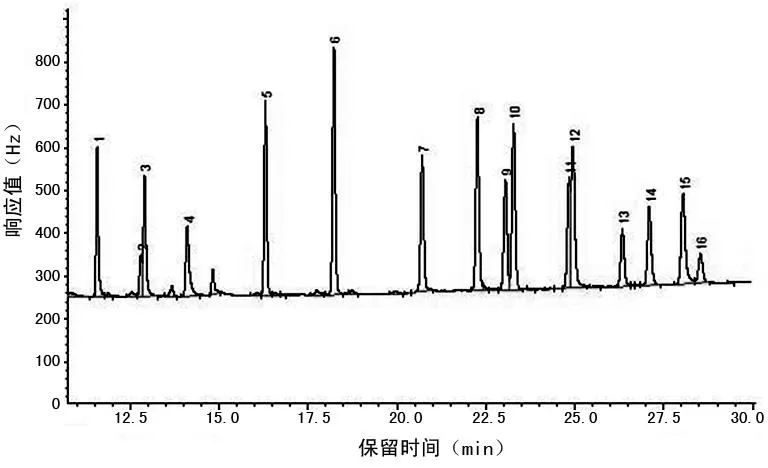
1~16:同圖1。
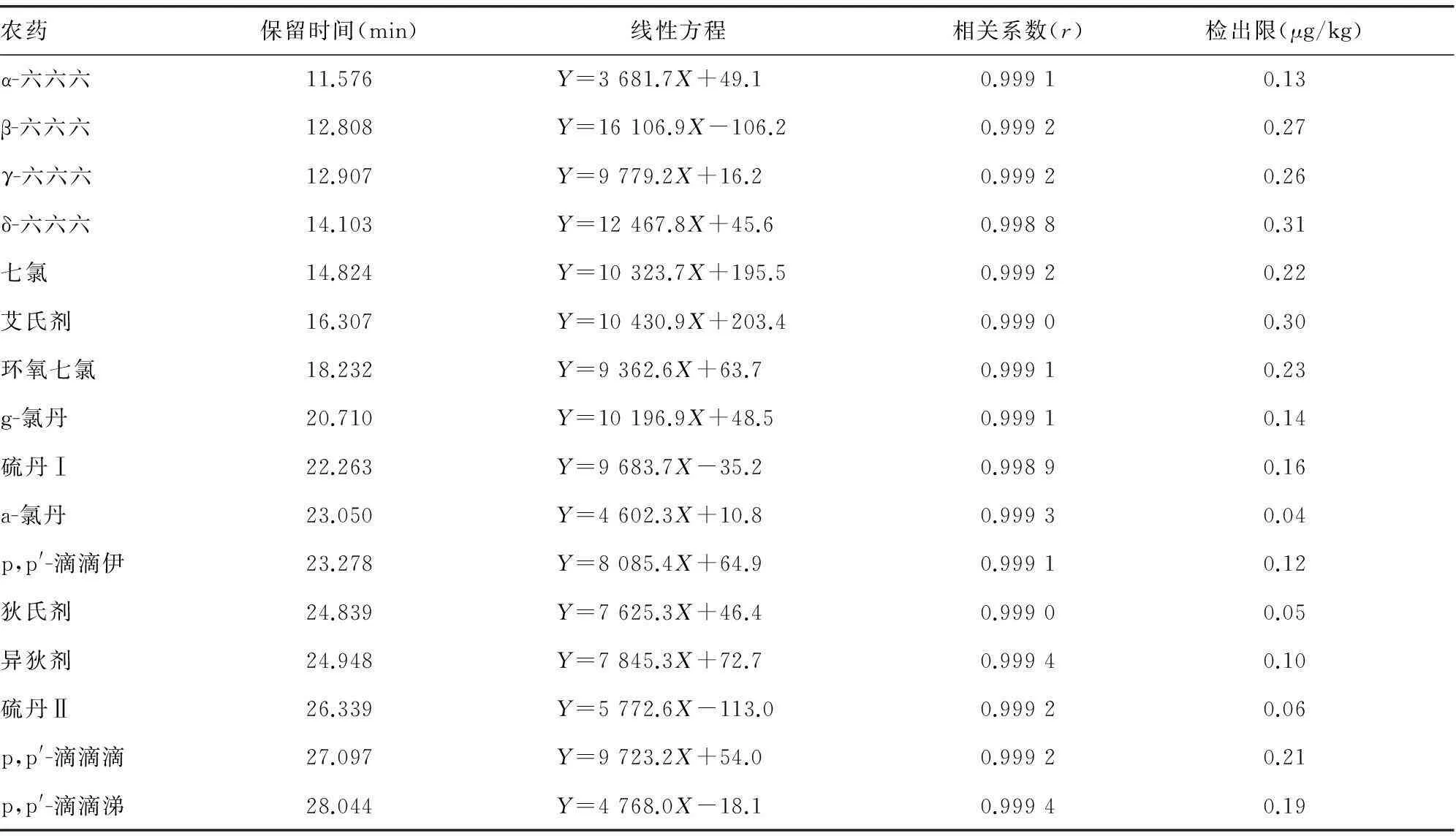
圖3 蟹肉基質加標(0.1 mg/L)的色譜圖
2.5精密度檢測和回收試驗分別取不含有上述16種農藥的空白水產品樣品作為基質,進行低、中、高3種不同水平(50、100、200 μg/kg)的加標回收試驗,每個水平平行檢測6次,3個加標水平的平均回收率為72.6%~115.2%,相對標準偏差(RSD)均小于7.5%,見表2(見《國際檢驗醫學雜志》網站主頁“論文附件”)。說明方法的準確度和精密度良好。
2.6樣品檢測應用本方法對40份不同采樣地點的水產品,如鯽魚、青蝦、螃蟹、河蚌等樣品進行有機氯農藥殘留的檢測,均未檢出有機氯。
3討論
目前,農藥多殘留分析方法采用的提取溶劑有丙酮、作為二氯甲烷、乙腈、正己烷等。通過試驗比較,本實驗采用乙腈作為溶劑,而乙腈也具有通用性強,對農藥的溶解度較大,可溶入油脂類雜質少,且分子小,組織穿透能力強等特點。
使用固相萃取柱可以有效凈化基質中脂肪等雜質,而不同的固相萃取柱對樣品基質凈化的效率也不相同。本文通過加標回收試驗比較了3種不同固相萃取柱的回收率,選擇Carb/NH2。
本文建立了采用乙腈提取,Carb/NH2固相萃取柱凈化,氣相色譜檢測水產品中有機氯類農藥殘留的檢測方法。本方法檢測靈敏度高,精密度好,線性好,能滿足有機氯類農藥殘留分析的要求,為水產品類食物中有機氯類農藥殘留分析提供了可靠的檢測手段,也為有機氯類及其他農藥品種的檢測提供了參考依據。
參考文獻
[1]Ameur WB,Trabelsi S,Megdiche YE,et al.Concentration of polychlorinated biphenyls and organochlorine pesticides in mullet (Mugil cephalus) and sea bass (Dicentrarchus labrax) from Bizerte Lagoon(Northern Tunisia)[J].Chemosphere,2013,74(3):2372-2380.
[2]田良良,史永富,王嬡,等.氣相色譜法測定蝦中有機氯農藥和多氯聯苯殘留量[J].分析試驗室,2014,33(9):1043-1046.
[3]張小輝,賈海燕,祁士華,等.漢江水體和魚體內有機氯農藥殘留水平及積累特征分析[J].安全與環境工程,2014,21(2):40-45.
[4]劉慧慧,徐英江,鄧旭修,等.萊州灣及東營近岸海域生物體中有機氯農藥和多氯聯苯污染狀況與風險評價[J].海洋與湖沼,2013,44(5):1325-1332.
[5]鄭林,施澤明,李佳宣,等.微波萃取氣相色譜法測定魚肉中有機氯殘留[J].四川理工學院學報:自然科學版,2010,23(1):62-64.
[6]張權,陳文生,洪亮,等.MSPD-GPC凈化GC-ECD法測定辣椒醬中8種有機氯農藥殘留分析[J].食品科學,2014,35(8):295-298.
[7]白有成,金海燕,盧勇,等.長江口嵊泗海域的生物體內持久性污染物殘留量及分布特征[J].海洋學研究,2011,29(3):162-168.
[8]安東,秦春艷,方展強,等.珠江口14種習見水生動物體內滴滴涕含量的測定與評價[J].天津農業科學,2014,20(1):33-39.
[9]肖麗和,刁璇,謝耀軒,等.QuEChERS法聯合在線GPC-GC-MS檢測冠心丹參膠囊中20種有機氯農藥殘留[J].中國新藥雜志,2014,23(15):1749-1753.
[10]孫曉杰,郭萌萌,王蘇玥,等.分散固相萃取-在線凝膠色譜-氣相色譜-質譜聯用法快速檢測紫菜中的農藥多殘留[J].色譜,2014,32(10):1124-1130.
[11]謝耀軒,王淑紅,王鐵杰,等.分散固相萃取-在線凝膠滲透色譜-氣相色譜-質譜法檢測香港中成藥中20種有機氯農藥殘留[J].沈陽藥科大學學報,2014,31(7):535-541.
[12]葉瑞洪,蘇建峰.分散固相萃取-超高效液相色譜-串聯質譜法測定果蔬、牛奶、植物油和動物肌肉中殘留的61種有機磷農藥[J].色譜,2011,29(7):618-623.

Determination of organochlorine pesticide in aquatic products by gas chromatography*1
SongXin1,2,HangXueyu2,WangQin1,2,WangLu2,FengXiaoqing2,XuRui2,MaoLi1△
(1.CollegeofPublicHealth,NanjingMedicalUniversity,Nanjing,Jiangsu211166,China;2.HuaianCenterforDiseaseControlandPrevention,Huaian,Jiangsu223001,China)
Abstract:ObjectiveA method for quantitative determination of 16 kinds of organochlorine pesticide (OCPs) in aquatic products by gas chromatography (GC)was established.MethodsThe sample was extracted by acetonitrile,purified by Carb/NH2,and then determined by GC.ResultsThe linear relations of 16 kinds of OCPs were good at 0.05-1.00 mg/L(r>0.998).The limit of detection of OCPs was in the range of 0.04-0.31 μg/kg(S/N=3).When the standard levels were 50,100,200 μg/kg,the recovery rates were 72.6%-115.2%,the relative standard deviations were 0.6%-7.5%.ConclusionThe method established in this study is applied to the determination of 16 kinds of OCPs in real samples with satisfactory results.
Key words:gas chromatography;aquatic products;organochlorine pesticide
(收稿日期:2015-12-20)
DOI:10.3969/j.issn.1673-4130.2016.06.006
文獻標識碼:A
文章編號:1673-4130(2016)06-0733-03
基金項目:淮安市科技局科技發展計劃基金項目(HAS2015030);2015年度淮安市疾控系統預防醫學科研課題項目 (hayf201520)。
作者簡介:宋鑫,男,主管藥師,主要從事理化檢驗研究。(△)通訊作者,E-mail:maoli@njmu.edu.cn。
猜你喜歡
中國纖檢(2016年12期)2017-01-20 09:28:19
現代農業科技(2016年20期)2016-12-20 14:51:09
現代農業科技(2016年20期)2016-12-20 09:05:36
分析化學(2016年7期)2016-12-08 00:09:44
分析化學(2016年7期)2016-12-08 00:07:08
價值工程(2016年29期)2016-11-14 01:34:54
科技視界(2016年24期)2016-10-11 18:58:00
考試周刊(2016年39期)2016-06-12 16:01:44
中國科技博覽(2016年4期)2016-04-25 07:25:47
中國科技博覽(2016年8期)2016-04-25 04:57:50
