白血病相關蛋白LRP16 過表達HepG2 穩定細胞系的構建及鑒定
2015-11-29 08:31:38李婷王安平李萍王雙雙母義明
生物技術通訊 2015年5期
關鍵詞:胰島素
李婷,王安平,李萍,王雙雙,母義明
解放軍總醫院 內分泌科,北京 100853
白血病相關蛋白16(leukemia related protein 16,LRP16)基因是韓為東等于1999年從正常人外周血淋巴細胞中克隆并命名的一個基因,屬于Macro Domain 家族。該基因定位于染色體11q12.23,含11個外顯子,編碼325 個氨基酸殘基構成的蛋白[1-2]。我們前期的研究表明,LRP16 基因對胰島素分泌和胰島素抵抗均有影響。它在脂肪細胞3T3-L1、肌肉細胞C2-C12、肝癌細胞HepG2 中發揮著重要作用,LRP16 過表達可上調多種炎性因子(TNF-α、IL-6)的表達,損傷IRS-1 信號傳導通路(降低pIRS-1、PI3-K、pAkt 的表達),抑制細胞胰島素刺激的葡萄糖攝取,引起外周胰島素抵抗[3-5]。在這些研究中我們采用的是質粒轉染的方式,這一技術一方面只能達到短期轉染,另一方面轉染效率較低。我們想深入研究LRP16 促進胰島素抵抗的分子機制,希望獲得長期穩定的LRP16 過表達細胞株。因此,我們構建了LRP16 過表達的慢病毒表達載體,且成功地感染了HepG2 細胞,獲得穩定過表達LRP16 的細胞株,為深入研究LRP16 抑制肝臟胰島素抵抗的相關機制提供相關技術支持。
1 材料和方法
1.1 材料
HEK293T 包裝細胞及人肝癌HepG2 細胞均購自中國醫學科學院基礎醫學研究所北京協和細胞資源中心(CRC/PUMC);過表達質粒載體EX-Y2069-Lv201 及對照質粒載體EX-NEG-Lv201(表1,圖1)由OmicsLink 公司構建;慢病毒包裝試劑盒Lenti-Pac HIV Expression Packaging Kit、Lenti-Pac HIV and FIV qRT-PCR 滴定試劑盒、第一鏈cDNA 合成試劑盒、2×All-in-One qPCR Mix 均購自Gene Copoeia 公司;MEM 培養基、DMEM 培養基、丙酮酸鈉、胎牛血清、青霉素-鏈霉素雙抗均購自Gibco 公司;TRIzol 購自Invitrogen 公司;蛋白marker 購自Thermo公司;Western 印跡發光試劑盒購自北京普利萊公司;鼠抗人β-actin抗體購自Cell Signaling Technology 公司;辣根過氧化物酶(HRP)標記的羊抗鼠IgG購自中杉金橋公司;qPCR 引物由Invitrogen 公司合成;LRP16抗體由本實驗室制備保存。
1.2 細胞復蘇與培養
選用25 mL 培養瓶,適量HepG2 細胞,加入5 mL MEM 培養基(含10%胎牛血清、1%青鏈霉素、1 μmol/L 丙酮酸鈉),于37℃、5% CO2培養箱中培養,2 d換液一次,4~5 d傳代一次。
1.3 質粒載體構建及鑒定
對構建的過表達質粒載體EX-Y2069-Lv201 的LRP16 開放讀框(ORF)進行測序,結果與人類LRP16基因(GenBank:NM_014067)序列相符。
1.4 慢病毒包裝
按照Lenti-Pac HIV Expression 慢病毒包裝試劑盒進行下述操作。在轉染前2 d,按每個10 cm的培養皿1.3×106~1.5×106細胞數將HEK293T 包裝細胞接種于培養皿中,用含10%胎牛血清的DMEM 培養基于37℃、5% CO2孵箱中培養,使細胞在轉染時達到70%~80%的密度;在去聚丙烯的試管中,取2.5 μg 病毒過表達質粒EX-Y2069-Lv201 和5.0 μL(0.5 μg/μL)Lenti-Pac HIV mix 稀釋至200 μL 的Opti-MEM 內;取15 μL EndoFectin Lenti,在另一試管中用Opti-MEM 稀釋至終體積為200 μL;將稀釋的EndoFectin Lenti 反應物加入輕微渦旋含DNA 的試管中(添加順序不可顛倒),然后將上述混合物轉移到5 mL 圓底聚丙烯試管Falcon 中;把形成的DNA-EndoFectin 復合物直接加入培養皿,輕輕搖晃培養皿使復合物均勻分布;在37℃、5% CO2孵箱中孵育細胞過夜(8~14 h),用含2%~5%經熱失活的胎牛血清和青霉素、鏈霉素的DMEM 培養基替換過夜的原培養基;加入1/500 體積的TiterBoost 反應物到培養基中,在37℃、5% CO2孵箱中繼續孵育;經48 h轉染,收集含假病毒的培養液到加蓋的試管中,500 r/min 離心10 min 去除細胞碎片;進一步離心,用0.45 μm 孔徑的濾器過濾HEK293T 細胞上清液,得到過表達病毒濃縮液LPP-Y2069-Lv201-400,分裝小管放置于-80℃備用。同樣步驟得到對照病毒濃縮液LPP-NEG-Lv201-100。

表1 載體克隆信息
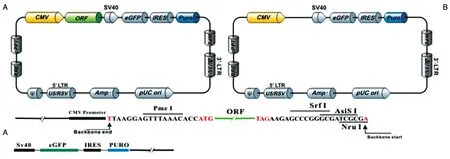
圖1 過表達質粒載體EX-Y2069-Lv201(A)及對照質粒載體EX-NEG-Lv201(B)信息
1.5 慢病毒滴度測定
HEK293T 細胞傳代,24 孔板中每孔加入1×105細胞,體積為500 μL;次日準備10 支無菌Ep 管,每管加入90 μL 培養基,取待測病毒原液10 μL 加入第1 支管中,混合均勻,取混合均勻的第1 管液10 μL 加入第2 支管中,繼續相同操作直到最后一管;選取所需細胞孔,吸去90 μL培養基,加入稀釋好的病毒溶液,于37℃、5% CO2培養箱中培養;1 d后,加入新鮮培養基500 μL;小心操作;4 d 后抽提RNA,然后采用Lenti-Pac HIV and FIV qRT-PCR 滴定試劑盒提取總RNA,將RNA 逆轉錄為cDNA,實時定量PCR檢測綠色熒光蛋白標記的慢病毒滴度。病毒的滴度等于帶有熒光的細胞數除以病毒原液量,即2/(1E-6)=2E+6 TU/μL,亦即2E+9 TU/mL。確保病毒滴度≥108TU/mL(LPP-Y2069-Lv201-400 為3.87×108TU/mL,LPP-NEG-Lv201-100 為6.25×108TU/mL),以便后續操作。
1.6 慢病毒載體感染HepG2細胞系
將HepG2 細胞計數后鋪到6 孔板中培養,各孔加入2 mL MEM 培養基(含10%胎牛血清,1%青鏈霉素,1 μmol/L 丙酮酸鈉),在37℃、5% CO2條件下培養24 h;鋪板24 h 后,按照MOI=30,每孔加入相應目的基因慢病毒液LPP-Y2069-Lv201-400 和對照慢病毒液LPP-NEG-Lv201-100,混勻后于37℃、5% CO2條件下培養;感染48 h 后,觀察慢病毒侵染結果,拍攝細胞熒光圖片;用胰酶消化感染過的細胞并轉移至6 孔板(每塊6 孔板對應一種慢病毒,留出1 孔用于陰性對照細胞),以含1 μg/mL 嘌呤霉素的MEM 培養基(含10%胎牛血清,1%青鏈霉素,1 μmol/L 丙酮酸鈉)進行藥物篩選培養5~7 d,觀察細胞存活情況和熒光圖片,當細胞熒光圖片與明場細胞相對應,則嘌呤霉素濃度可以減半進行培養;連續加藥培養7 d后,嘌呤霉素濃度減半至0.5 μg/mL 繼續培養,待細胞長滿后收取細胞。
1.7 qPCR檢測LRP16的mRNA表達
從細胞中提取RNA,定量,電泳確定其純度。用第一鏈cDNA 合成試劑盒進行逆轉錄,用Gene Copoeia 公司試劑盒配置所推薦的反應體系后在PRISM 7300(ABI 公司)上進行qPCR 反應。根據LRP16 的基因序列設計PCR 引物(上游引物:5'-A GCTCTTTCACTCGGAGGATT-3';下游引物:5'-TG GCAAAAGAAGAAGACAAAGC-3')。以GAPDH 基因為內參,根據GAPDH 的基因序列設計PCR 引物(上游引物:5'-TGGGTGTGAACCACGAGAA-3';下游引物:5'-GGCATGGACTGTGGTCATGA-3')。反應條件:95℃ 30 s 預變性;95℃ 5 s,60℃ 20 s,72℃10 s,40 個循環。目標基因mRNA 的相對含量通過ΔΔCt計算方法獲得。
1.8 Western印跡檢測LRP16的表達
用自制IP 緩沖液(0.15 mol/L NaC1,20 mmol/L Tris-HC1,10%甘油,0.1% NP-40)加入25×總蛋白酶抑制劑及用100×PMSF 配置的細胞裂解液,分別裂解過表達穩轉株、對照穩轉株和野生型(未轉染)HepG2 細胞,4℃、12 000 r/min 離心后取上清作為樣品,測定蛋白濃度,取等量蛋白樣品進行SDSPAGE 后轉印蛋白至PVDF 膜,用以TBST 液配制的5%脫脂奶粉于室溫封閉2 h,LRP16 鼠抗以1∶200稀釋,β-actin 鼠抗人以1∶1000 稀釋,4℃反應過夜,用TBST 液洗膜3 次,每次15 min,二抗以1∶5000 稀釋,室溫反應1 h,用TBST液洗膜3次,每次15 min,在膜上滴加發光液后進行曝光。
2 結果
2.1 慢病毒載體感染HepG2細胞株的篩選
按前述方法將HepG2 細胞計數后鋪到6 孔板中培養,每孔加入目的基因慢病毒液LPP-Y2069-Lv201-400 和對照慢病毒液LPP-NEG-Lv201-100,感染48 h 后,觀察慢病毒侵染結果,拍攝細胞熒光圖片(圖2)。可以看出,轉入LPP-Y2069-Lv201-400 和LPP-NEG-Lv201-100 慢病毒后的HepG2 細胞,其綠色熒光蛋白表達超過90%。
2.2 過表達慢病毒感染HepG2 細胞后對LRP16 基因表達的影響
提取過表達穩轉株、對照穩轉株和野生型(未轉染)HepG2 細胞總RNA,采用實時熒光定量PCR 檢測各組細胞的LRP16 相對表達量。結果顯示,過表達穩轉株LPP-Y2069-Lv201-400 中LRP16 目的基因的表達量為對照穩轉株LPP-NEG-Lv201-100 的128.64倍(表2)。
2.3 HepG2 細胞的感染和Western 印跡分析LRP16蛋白的過表達效果
將獲得的LRP16 基因過表達的HepG2 細胞、過表達對照細胞及野生型HepG2 細胞分別培養4~5 d,在培養皿中加入適量細胞裂解液提取蛋白,用上清液行SDS-PAGE,轉膜后進行蛋白免疫印跡分析。結果顯示空白組和對照組細胞在相對分子質量約28×103處均出現了微弱條帶,而過表達組出現明顯的粗黑條帶,表明過表達穩轉株LPP-Y2069-Lv201-400 的LRP16 表達量明顯高于對照穩轉株LP-NEG-Lv201-100和野生型HepG2(圖3),結果與實時熒光定量PCR一致。
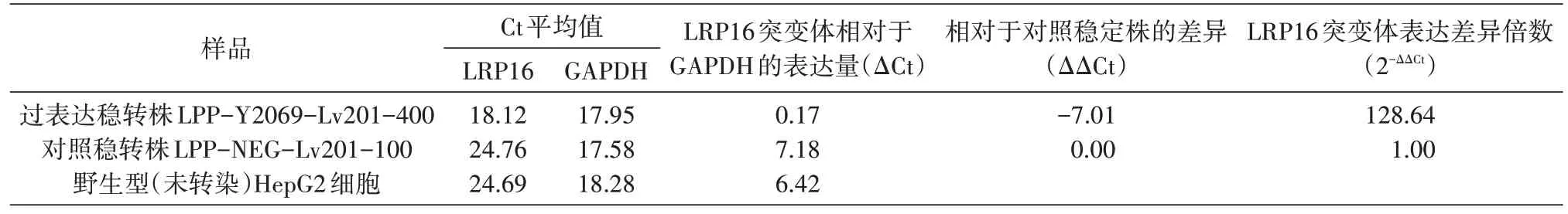
表2 qPCR檢測過表達穩轉株、對照穩轉株及野生型HepG2細胞中LRP16 mRNA水平
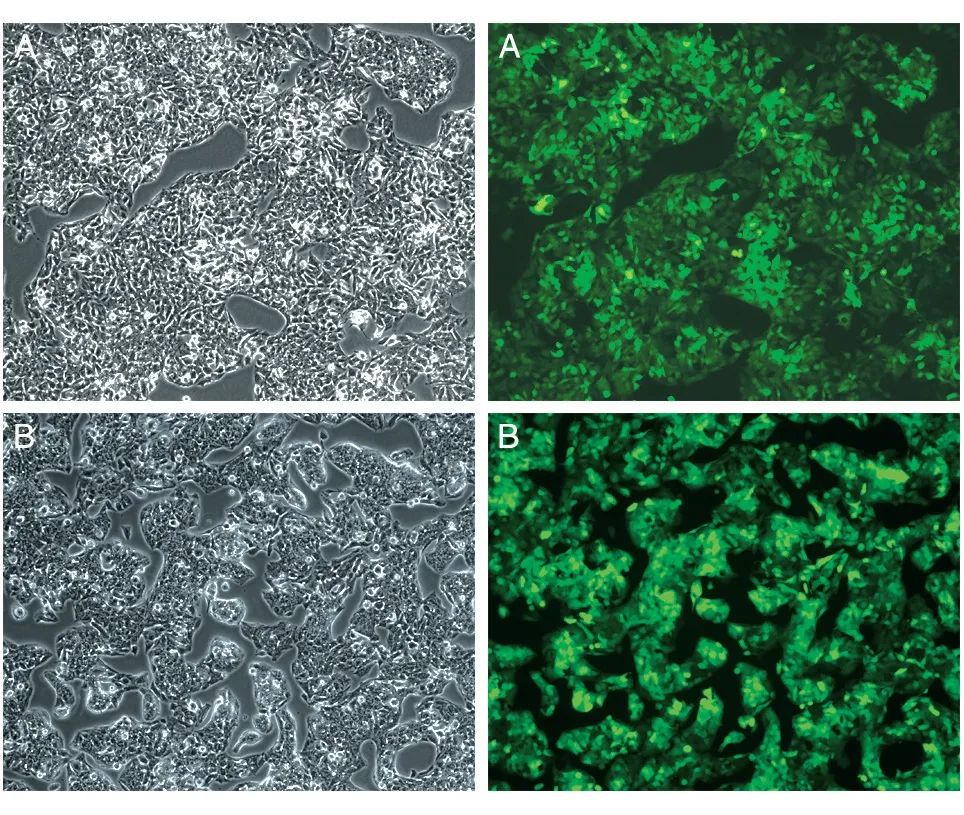
圖2 LRP16過表達穩定株(A)及對照穩定株(B)熒光轉染情況

圖3 Western印跡檢測LRP16蛋白表達的差異
3 討論
前期,我們做了大量LRP16相關研究,發現它是機體發育過程中不可或缺的一個基因,其表達不僅與腫瘤的發生發展密切相關[6-7],也參與了胰島素的分泌及胰島素外周器官的胰島素抵抗作用。
隨著糖尿病發生率的上升,胰島素抵抗一度成為近年的研究熱點,胰島素通路受到重視。而我們發現的LRP16 基因參與胰島素抵抗立刻引起了關注。但目前我們的發現大多停留在表明現象,真正所涉及到的機制研究甚少。
因為肝臟是胰島素發揮作用的重要外周器官,而且之前我們也曾在肝癌HepG2 細胞系中研究過LRP16的作用,所以,在深入研究LRP16基因對胰島素信號通路的影響機制時,我們仍然選用這一常用的肝癌細胞系。此LRP16 基因過表達的HepG2 穩定細胞系的建立,為我們今后的研究提供了一個長期有效的研究工具。
[1]韓衛東,于力,樓方定,等.一個新的白血病相關基因LRP16全長cDNA 的克隆、序列分析及表達特征[J].中國生物化學與分子生物學報,2001,17(2):209-204.
[2]于力,韓衛東,樓方定,等.新的白血病相關基因LRP16 的克隆[J].軍醫進修學院學報,2000,21(2):81-84.
[3]臧麗,呂朝暉,汪保安,等.LRP16 通過下調PPARγ表達抑制細胞葡萄糖攝取[J].中華內分泌代謝雜志,2010,26(3):217-220.
[4]臧麗,母義明,呂朝暉,等.LRP16基因抑制IRS-1信號通路和過氧化物酶體增殖物激活受體γ轉錄活性導致C2-C12 成肌細胞胰島素抵抗[J].中華醫學雜志,2011,91(20):1408-1412.
[5]臧麗,母義明,呂朝暉,等.人白血病相關蛋白16 基因對3T3-L1脂肪細胞葡萄糖攝取及過氧化物酶體增殖物激活受體γ活性的影響[J].中國糖尿病雜志,2011,19(4):293-297.
[6]Meng Y G,Han W D,Zhao Y L,et al.Induction of the LRP16 gene by estrogen promotes the invasive growth of Ishikawa human endometrial cancer cells through the downregulation of E-cadherin[J].Cell Res,2007,17:869-880.
[7]Tian L Y,Wu Z Q,Zhao Y L,et al.Differential induction of LRP16 by liganded and unliganded estrogen receptor alpha in SKOV3 ovarian carcinoma cells[J].J Endocrinol,2009,202:167-177.
猜你喜歡
人人健康(2023年26期)2023-12-07 03:55:46
家庭醫藥(2019年9期)2019-09-23 18:54:32
中國生殖健康(2019年2期)2019-08-23 08:12:10
家庭科學·新健康(2018年8期)2018-10-30 10:23:20
人生與伴侶·共同關注(2018年5期)2018-08-15 10:00:00
科學生活(2016年9期)2016-10-20 13:12:45
中國衛生標準管理(2015年1期)2016-01-14 03:41:27
人人健康(2015年17期)2015-09-09 16:25:20
藥學與臨床研究(2015年4期)2015-06-05 11:35:51
中國醫藥科學(2015年15期)2015-02-27 12:32:27
