PAC投加對絮體破碎后再絮凝特性和顆粒去除的影響
2015-09-21 01:41:04赫俊國何開帆董志虎袁一星
哈爾濱工業大學學報 2015年2期
赫俊國,劉 劍,何開帆,于 淼,董志虎,袁一星,2,張 杰,2
(1.哈爾濱工業大學市政環境工程學院,150090哈爾濱;2.城市水資源與水環境國家重點實驗室(哈爾濱工業大學),150090哈爾濱)
給水處理過程中,混凝是去除水中膠體或懸浮顆粒最常用的方法,也是關鍵的工藝步驟之一[1].混凝過程一般分為凝聚和絮凝兩部分,凝聚包括金屬離子水解、粒子遷移及顆粒脫穩等一系列過程,而絮凝是脫穩后的膠體或微小懸浮物逐漸成長為較大絮凝體的過程[2-3].混凝的目的是使膠體脫穩和脫穩后的膠體-絮體達到一定的尺寸和強度,從而提高膠體及懸浮顆粒的去除效果.合理地進行絮凝工藝設計,必須保證各構筑物單元內絮體破碎率最小化.在實際生產過程中,具有較大剪切強度的區域普遍存在于給水處理廠各構筑物單元[4].當絮體暴露在較高強度的剪切力條件下時會被打碎[5],打碎后形成的小顆粒在沉淀池去除率降低,從而影響后續工藝的處理效能[6-7].
破碎可以分為表面破損和大尺度破碎兩種模式[8].前者是指微小的顆粒從絮體表面掉落,致使微小絮體的數量增加;后者是指大絮體分裂為具有相似大小的絮體,微小絮體的數量并沒有明顯增多.目前,針對絮體破碎再絮凝的研究主要集中于不同混凝劑種類形成的絮體,在不同破碎強度下破碎后能否恢復到原有水平.Wang等[9-10]對氯化鋁(AlCl3)和聚合氯化鋁(PAC)形成的絮體破碎再絮凝過程進行了研究,認為破碎后的絮體發生再絮凝時,絮體大小很難恢復到破碎前的水平.Xu 等[11]對 Al13、PAC 與腐殖酸形成的絮體的研究結果表明,兩種藥劑形成的絮體破碎再絮凝過程均不可逆,Al13絮體強度小于PAC絮體,但Al13絮體的恢復能力強于PAC絮體.Jarvis等[12-13]認為Fe-高嶺土絮體及Fe-天然有機物(NOM)絮體破碎后的再絮凝能力較差,絮體大小不能恢復到未破碎前的水平.然而,部分研究表明,絮體破碎-再絮凝過程完全可逆.Lin等[14]認為高嶺土與陽離子淀粉形成的絮體的破碎再絮凝過程完全可逆,同時,中性或堿性條件下形成的絮體的抗剪切能力強于酸性條件下形成的絮體.Yukselen等[15-16]對正電荷聚合電解質進行了研究,認為其在長時間攪拌條件下形成的絮體破碎后基本能恢復到未破碎前的大小.Yu等[17-18]對不同混凝機理條件下絮體破碎與再絮凝進行了研究,認為電中和條件下形成的絮體的破碎再絮凝過程可逆,而網捕卷掃條件下形成的絮體破碎再絮凝過程不可逆.目前,對不同混凝劑形成的絮體的破碎再絮凝過程研究較多,而關于如何提高破碎再絮凝過程不可逆絮體再絮凝能力的研究較少,且對再絮凝階段絮體的生長速率變化也未進行系統研究.同時,如何改善低溫低濁水條件下形成的絮體破碎后的沉降性能鮮有報道.本文以低溫低濁水為處理對象,以PAC為混凝劑,分別考察了再絮凝階段補投PAC對絮體粒徑、生長速率、粒徑分布變化,濁度及顆粒數去除效果的影響,以期改善絮體破碎后的再絮凝能力,提高顆粒的去除效率,從而為低溫低濁水的混凝處理提供技術支持.
1 實驗
1.1 實驗原水及材料
實驗原水來自青海省西寧市第七水廠,該水廠原水為雪山融化水,水質具有較好的穩定性.實驗在2012年11月~2013年3月進行,實驗期間原水水質:水溫為1.4~4.3℃,濁度為 1.89~1.96 NTU,pH 為 8.01~8.07,顆粒數為 367~389個·mL-1,顆粒粒徑為 1.12~1.78 μm.
PAC(化學純)為實驗所在水廠使用的混凝劑,鹽基度為40%~90%,鋁質量分數(以氧化鋁計)≥30%.每次實驗前2 h,將PAC溶于蒸餾水,配成質量分數為5%的使用溶液.
1.2 實驗方法
絮凝-破碎-再絮凝實驗用六聯攪拌器(MY3000-6M,中國武漢梅宇儀器設備有限公司)完成.將1 L的實驗原水置于1 L的圓形燒杯中,并按以下條件進行絮凝實驗:依據水廠現狀及前期藥劑優選實驗,首先將15 mg/L的PAC使用溶液投加到燒杯中,以300 r/min的轉速快速攪拌0.5 min,然后以120 r/min的轉速攪拌1.5 min,最后以70 r/min的轉速慢攪6 min.絮凝結束后,立即進行破碎實驗,破碎條件為:轉速500 r/min,時間1 min.當破碎實驗結束后,向燒杯中再次投加不同量的PAC使用溶液,并立即進行再絮凝實驗,再絮凝攪拌強度為70 r/min,攪拌時間為1~6 min,并沉淀20 min.分別利用濁度儀(2100P,美國哈希公司)和在線顆粒計數儀(2200PCX,美國哈希公司)測定不同再絮凝時間沉后水上清液中的剩余濁度和剩余顆粒數.
利用激光粒度儀(Mastersizer2000,英國馬爾文儀器公司)對絮凝、破碎、再絮凝過程的粒徑變化進行在線監測[19],實驗中粒徑均為絮體的平均粒徑[20].實驗過程中,在蠕動泵的作用下,圓形燒杯中的原水以2.0 L/h的速度緩慢地進入粒度儀,儀器中的光檢測單元可以測定絮體的粒徑,經過檢測后的水樣回流至燒杯[7].再絮凝過程中絮體粒徑分布同樣采用激光粒度儀進行監測[21].
2 結果與分析
2.1 絮體粒徑的變化
絮凝過程中,絮體粒徑可以間接反映絮體的沉降性能,一般大絮體沉降性好,小絮體沉降性差;同時,絮凝、破碎及再絮凝過程粒徑的變化可以直觀地表征絮體形態的變化.不同PAC補投量時絮凝、破碎及再絮凝過程絮體粒徑變化如圖1所示.

圖1 不同PAC補投量時絮體粒徑的變化
由圖1可以看出,絮凝過程可以分為3個階段:停滯期、快速增長期和穩定期.停滯期,混凝劑與原水進行充分混合,其水解產物與原始顆粒發生反應,在此階段,絮體大小變化甚微.快速增長期,隨著反應時間的延長,絮體粒徑迅速增大,當絮凝時間從1 min延長至6 min時,絮體粒徑由17 μm增大到370 μm.在此階段,原始顆粒表面所帶的負電荷被PAC所帶的大量正電荷強烈中和,粒子之間相互碰撞形成較大的絮體;同時,由于PAC混凝劑的網捕架橋作用,可以將較小的顆粒吸附在大顆粒上,從而使絮體尺寸不斷增大.絮凝時間超過6 min后,繼續延長絮凝時間,絮體大小幾乎不發生變化,此時絮體生長已處于穩定期,在該階段,絮凝過程為絮體生長和破碎的動態平衡過程.
破碎開始后,絮體粒徑急劇減小.破碎0.5 min時,絮體粒徑由 370 μm 下降到 176 μm.繼續延長破碎時間,絮體粒徑繼續減小,破碎完成時的絮體粒徑為109 μm.由于絮體極易破碎,當作用于絮體表面的剪切力大于絮體的內部結合力時,絮體將發生破碎,粒徑下降.破碎后的絮體粒徑小、沉降性能差,在沉淀過程難以去除,從而影響后續工藝的處理效能.
再絮凝階段,在混凝劑補投與不補投的條件下,絮體粒徑均隨著攪拌時間的增加逐漸增大,說明PAC絮體破碎后具有一定的再絮凝能力.再絮凝階段并沒有出現停滯期,這可能與破碎后絮體的表面活性有關.絮凝階段,當向水中投加混凝劑后,微小絮體在電中和、網捕卷掃作用下快速凝聚,當絮體發生破碎后,絮體內部的結合鍵斷裂并暴露在外,絮體活性并沒有消失.當再次投加PAC后,破碎后的絮體能夠很快地發生碰撞,絮體粒徑迅速增大.不補投混凝劑時,絮體粒徑變化非常緩慢,再絮凝結束時,絮體粒徑為225 μm,僅為破碎前的59.5%,說明PAC絮體破碎后不能完全恢復.隨著混凝劑補投量的增大,絮體的再絮凝能力呈現先升高后下降的趨勢,再絮凝末期絮體粒徑也先增大后減小.PAC補投量較低時(1 mg/L),絮體粒徑不能完全恢復到破碎前的水平;補投量增大到2 mg/L時,絮體粒徑由118 μm增大到400 μm,明顯大于未破碎前的 370 μm,絮體粒徑增長率為8.11%;補投量為4 mg/L時,絮體粒徑達到最大,為408 μm,但相較于2 mg/L時的增加幅度并不明顯.當絮體破碎后,絮體表面的電荷發生變化,且分布更加均勻[10].隨著投藥量的升高,PAC表面所帶的大量電荷會與裸露在外的負電荷發生中和反應;由于PAC的網捕卷掃作用,破碎后的小絮體迅速碰撞并形成大絮體.同時,隨著投藥量的升高,體系中發生電中和作用和網捕卷掃作用的載體不斷升高,更多的微小絮凝體逐漸形成較大的絮體,故絮體粒徑逐漸增大.當投藥量為4 mg/L時,體系中的原有電荷基本被中和,故投藥量的增加對絮體粒徑增長影響不大.繼續增加投藥量,絮體的粒徑反而減小.補投量為8 mg/L時,絮體粒徑減小到345 μm,小于破碎前的粒徑.這是因為在初始絮凝過程中,已經投加了大量的PAC,絮體表面的電荷已被中和,雖然破碎后絮體表面的電荷進行了重新分布,但當PAC的投加量過大時,小絮體發生再穩,阻礙了微小絮體的相互碰撞,致使絮體粒徑變小.
2.2 再絮凝過程絮體生長速率的變化
隨著PAC補投量的升高,再絮凝階段絮體粒徑達到平衡粒徑所需的時間不斷縮短(圖1).不補投PAC時,絮體再絮凝的整個過程均在生長;投加量為1,2 mg/L時,粒徑達到平衡所需時間分別為4.5,3.5 min;較大補投量(4~8 mg/L)時,絮體粒徑達到穩定所需時間基本維持在2.5 min.由圖1還可以看出,再絮凝階段絮體生長過程中,粒徑與絮凝時間基本呈線性關系.因此,對絮體生長曲線進行線性擬合,擬合直線的斜率即為再絮凝過程絮體的生長速率,結果如表1所示.
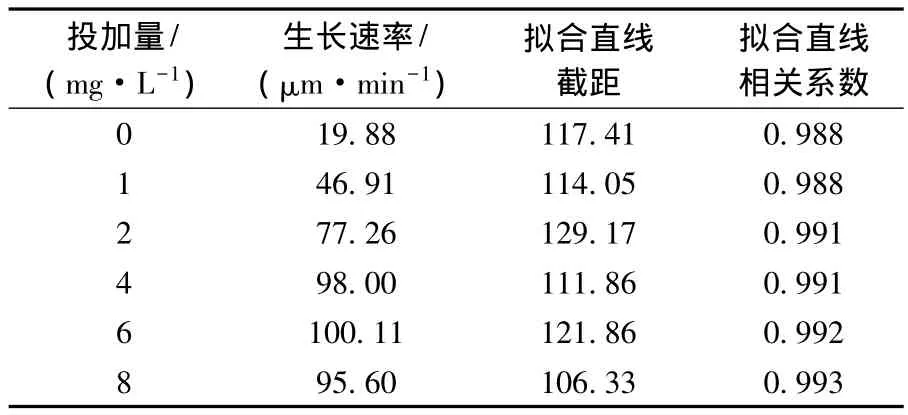
表1 PAC再次投加量對絮體生長速率的影響
不同補投量時,絮體粒徑與絮凝時間之間存在良好的線性關系,相關系數均達0.98.代表破碎后絮體尺寸的擬合直線截距基本相同,維持在106~129,說明實驗的重復性較好,這與絮凝-破碎條件一致有關.隨著PAC補投量的升高,再絮凝速率呈現先增加后降低的趨勢.不補投PAC時,絮體破碎后的再絮凝速率為19.88 μm/min,補投少量(1 mg/L)的 PAC時,再絮凝速率為46.91 μm/min,增加了 2.4倍,說明補投少量的PAC即可加快絮體破碎后的再絮凝速率.當PAC補投量為2 mg/L時,再絮凝速率提高到77.26 μm/min.PAC補投量為6 mg/L時,速率達到最大,為 100.11 μm/min,繼續提高補投量,再絮凝速率緩慢下降.隨著補投量的升高,與顆粒發生反應的電荷數量不斷增多,且參與網捕卷掃作用的官能團數量不斷提高,從而提高了絮體的生長速率[22].當補投量過大時(8 mg/L),絮體的再穩使得再絮凝速率下降.對比圖1、表1及圖3可得,PAC為6 mg/L時,絮體破碎后的再絮凝速率達到最大,但再絮凝結束后的絮體粒徑并非最大,且此時的剩余濁度較高,說明生長速率的快慢只是反映絮凝好壞的一個因素,不能單純以再絮凝速率來比較絮體破碎后的再絮凝能力,應綜合考慮絮體的粒徑及對顆粒的去除能力.
2.3 絮體粒徑分布的變化
為分析絮體生長過程中不同粒徑顆粒的遷移變化規律,分別考察了PAC補投量為0及2 mg/L時,再絮凝階段絮體粒徑分布的變化,結果如圖2所示.
不補投混凝劑時,隨著絮凝時間的延長,絮體粒徑分布曲線逐漸向大粒徑的方向移動,但移動速度較慢(圖2(a)).當絮體發生破碎后,絮體粒徑分布曲線呈現雙峰,說明絮體破碎后微小絮體的數量急劇增多.絮體粒徑分布曲線雙峰對應的粒徑大小分別為22和115 μm,所占體積分數分別為2.98%和7.63%.由圖2(a)可得,粒徑在22 μm附近的微小絮體的粒徑分布范圍較寬,而粒徑在115 μm附近絮體的粒徑分布范圍較窄,說明絮體破碎后,系統中以微小粒徑的絮體為主.當絮凝時間為2 min時,粒徑分布曲線仍有雙峰,但微小絮體的數量明顯減少,較大的絮體在不斷形成.雙峰所對應的絮體粒徑分布為31和178 μm,且粒徑為31 μm的絮體所占的體積分數為1.91%,明顯小于0 min時小粒徑峰值所占的體積分數.當再絮凝時間大于4 min時,粒徑分布曲線上的雙峰消失,峰值所對應的粒徑逐漸增大.再絮凝結束時,粒徑分布范圍由破碎時的1.12~447 μm變為再絮凝后的1.78~1 588 μm.由圖2(a)還可以看出,隨著絮凝時間的增加,粒徑分布峰值對應的絮體所占的體積分數逐漸減少,絮凝時間由2 min增加到6 min時,峰值所占百分比由7.35%降低到6.57%,說明當無混凝劑補投時,絮體粒徑范圍隨著絮凝時間的增加而變寬,絮體的粒徑分布更加分散.
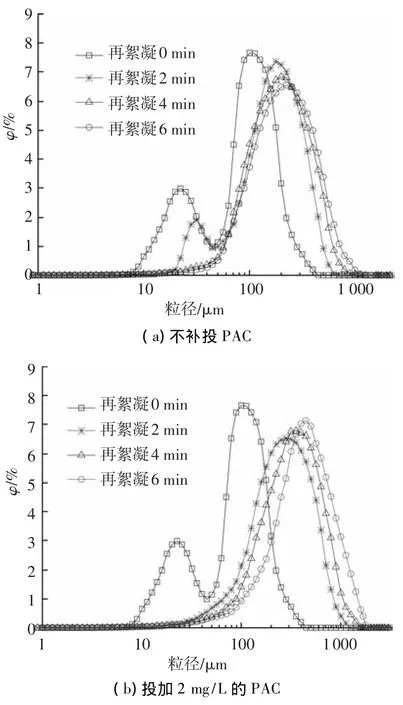
圖2 補投PAC對再絮凝過程粒徑分布的影響
補投2 mg/L PAC時,隨著絮凝時間的增加,絮體粒徑分布同樣向大粒徑的方向移動,但移動速度明顯加快(圖2(b)).再絮凝2 min時,絮體粒徑分布曲線上的雙峰已經消失,峰值對應的絮體粒徑增大到282 μm,明顯大于無投藥時的178 μm.隨著絮凝時間的繼續增加,粒徑分布曲線峰值對應的絮體粒徑也不斷增大,再絮凝結束時,峰值對應的粒徑為400 μm,且絮體粒徑分布范圍為4~1 782 μm,說明大粒徑絮體的數量明顯增多.圖2(b)表明,隨著絮凝時間的增加,粒徑分布曲線逐漸變窄,說明絮體的粒徑分布更加緊密、均勻.同時,粒徑分布曲線峰值對應的絮體所占的體積分數逐漸升高,由2 min時的6.55%升高到6 min時的7.13%,進一步說明大絮體的數量不斷增加.導致上述現象的原因可能是,當再次投加PAC后,破碎后形成的小絮體相互碰撞,逐漸形成較大粒徑的絮體,同時由于PAC的吸附架橋及網捕卷掃作用,大部分微小絮體被吸附進入大絮體的孔隙中,從而使系統中小顆粒數量變少,絮體的粒徑分布更加狹窄、均勻[23].
比較圖2(a)、(b)可得,當補投2 mg/L PAC后,粒徑范圍為280~630 μm的大絮體所占體積分數由22.85%升高到46.36%,而粒徑范圍為4~35 μm的小絮體所占體積分數由1.21%降低到0.19%,說明投加PAC能明顯減少小絮體的體積分數,增加大絮體的數量,從而提高絮體的再絮凝能力.
2.4 剩余濁度的變化
濁度是考察給水處理工藝運行好壞最直接的指標.圖3為不同PAC補投量條件下,沉后水剩余濁度隨再絮凝時間的變化.
不同PAC補投量條件下,沉后水上清液剩余濁度均隨再絮凝時間的增加而不斷降低.補投量為0時,剩余濁度隨時間的變化非常緩慢.絮凝6 min后,剩余濁度由再絮凝前的2.74 NTU降到再絮凝后的2.32 NTU,濁度去除率僅為15.33%.這是因為在絮體破碎過程中,絮體表面活性降低,影響了絮體的再絮凝能力,系統中微小顆粒不能被沉淀去除,從而導致濁度較高[24].補投PAC后,剩余濁度明顯降低,隨著再絮凝時間的增加,剩余濁度快速下降.當投藥量為2 mg/L時,沉后水剩余濁度為0.79 NTU,完全能夠滿足生活飲用水水質標準對濁度的要求;當絮凝時間為5 min時,剩余濁度小于1 NTU,說明再次投加PAC可以明顯提高系統對濁度的去除能力.上述現象的產生可能與破碎后絮凝體的表面特性有關,由于PAC中含有部分與再絮凝能力有關的單體鋁(Ala),當絮凝體發生破碎后,體系中的Ala分布在絮凝體的表面,從而提高了絮體的再絮凝能力[25].當 PAC 補投量為 4 mg/L,沉后水剩余濁度變化并不明顯.當PAC補投量大于等于6 mg/L時,沉后水剩余濁度反而比低投藥量時高,這一結果與圖1中粒徑的變化趨勢一致.
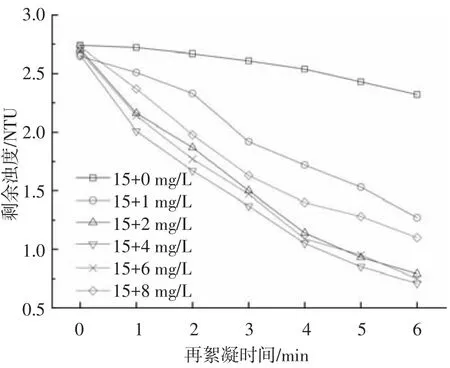
圖3 再次投加PAC對濁度去除效能的影響
2.5 剩余顆粒數的變化
剩余顆粒數可以直觀反映水中雜質顆粒的含量.不同PAC補投量條件下,再絮凝不同時間后的沉后水剩余顆粒數的變化如圖4所示.可以看出,隨著再絮凝時間的增加,沉后水剩余顆粒數不斷降低,但顆粒數降低的速率與投藥量有關.不補投PAC時,剩余顆粒數的變化非常緩慢,再絮凝結束時,沉后水剩余顆粒數為275個·mL-1,去除率僅為19.35%.該現象說明PAC絮體發生破碎后,產生了大量的微小絮體顆粒,該部分顆粒在再絮凝過程中不能有效地凝結為大絮體,沉降性能降低.再次投加PAC后,隨著投藥量的升高,沉后水剩余顆粒數呈現先降低后升高的趨勢.補投量較低時(1 mg/L),顆粒數有較高的去除效率,再絮凝結束后,沉后水剩余顆粒數為145個·mL-1,去除率為56.06%.再次投加PAC后,由于其較強的電中和能力及網捕卷掃能力,可以很好地將微小絮體顆粒凝結成較大的絮體,從而降低了系統中顆粒的數量.提高PAC的補投量,剩余顆粒數進一步降低,當補投量為2 mg/L時,剩余顆粒數降低到90個·mL-1,去除率為73.45%,繼續增加混凝劑的補投量,剩余顆粒數降低并不明顯.當PAC補投量較高時(>6 mg/L),剩余顆粒數隨投藥量的升高也不斷提高,這可能是過量的PAC使得絮體表面的電荷發生轉換,增大了顆粒間的排斥力,阻礙了顆粒間的有效碰撞,進而導致剩余顆粒數增多.
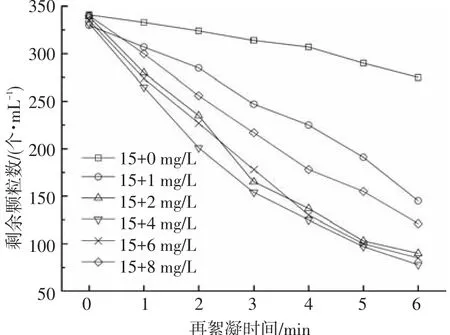
圖4 再次投加PAC對出水剩余顆粒的影響
比較圖3、4可知,沉后水剩余濁度與剩余顆粒數的變化規律基本相似,說明水中濁度與顆粒數具有一定的相關性.由此可得,濁度和顆粒數均可用于表征原水中膠體及雜質顆粒的含量.
3 結論
1)再次投加PAC可以明顯改善絮體破碎后的再絮凝能力,增大絮體粒徑及生長速率,提高濁度及顆粒數的去除效果,降低沉后水濁度及顆粒數.
2)隨PAC補投量的升高,絮體的粒徑先增大后減小,投藥量為4 mg/L時,粒徑達到最大為408 μm;絮體生長速率先升高后降低,生長速率達到最大值100.11 μm/min所需投藥量為6 mg/L.
3)當PAC補投量從0升高到2 mg/L時,大絮體明顯增多,小絮體顯著降低,280~630 μm絮體顆粒由22.85%增大到46.36%,4~35 μm絮體顆粒由1.21%減小至0.19%.
[1]SUN J,QIN L,LI G S,et al.Effect of hydraulic conditions on flocculation performances and floc characteristics in Chinese herbal extracts by chitosan and chitosan hydrochloride[J].Chemical Engineering Journal,2011,225:641-649.
[2]TAMBON, WATANABEY.Physicalaspect of flocculation — I:fundamental treatise[J].Water Research,1979,13:429-439.
[3]XIAO F,HUANG J C H,ZHANG B J,et al.Effects of low temperature on coagulation kinetics and floc surface morphology using alum[J].Desalination,2009,237:201-213.
[4]JARVIS P,JEFFERSON B,GREGORY J,et al.A review of floc strength and breakage [J].Water Research,2005,39:3121-3137.
[5]LI T,ZHU Z,WANG D S,et al.The strength and fractal dimension characteristics of alum-kaolin flocs[J].International Journal of Mineral Processing,2007,82:23-29.
[6]YU W Z,GREGORY J,CAMPOS L.The effect of additional coagulant on the re-growth of alum-kaolin flocs [J].Separation and Purification Technology,2010,74(3):305-309.
[7]WEI J C,GAO B Y,YUE Q Y,et al.Strength and regrowth properties of polyferric-polymer dual-coagulant flocs in surface watertreatment[J].Journalof Hazardous Materials,2010,175:949-954.
[8]MIKKELSEN L H,KEIDING K.The shear sensitivity of activated sludge:an evaluation of the possibility for a standardised floc strength test[J].Water Research,2002,36:2931-2940.
[9]WANG Y,GAO B Y,XU X M,et al.Characterization of floc size,strength and structure in various aluminum coagulants treatment[J].Journal of Colloid and Interface Science,2009,332:354-359.
[10]MCCURDY K,CARLSON K,GREGORY D.Floc morphology and cyclic shearing recovery:comparison of alum and polyaluminium chloride coagulants [J].Water Research,2004,38:486-494.
[11]XU W Y,GAO B Y,YUE Q Y,et al.Effect of shear force and solution pH on flocs breakage and re-growth formed by nano-Al13polymer[J].Water Research,2010,44:1893-1899.
[12]JARVIS P,JEFFERSON B,PARSONS S.Breakage,regrowth,and fractal nature of natural organic matter flocs[J].Environmental Science and Technology,2005,39:2307-2314.
[13]JARVIS P,JEFFERSON B,PARSONS S.The duplicity of floc strength [J].Water Science and Technology,2004,50(12):63-70.
[14]LIN Q T,PENG H L,LIN Q L,et al.Formation,breakage and re-formation of flocs formed by cationic starch [J].Water Science and Technology,2013,68(6):1352-1358.
[15]YUKSELEN M A,GREGORY J.The effect of rapid mixing on the break-up and re-formation of flocs[J].Journal of Chemical Technology and Biotechnology,2004,79:782-788.
[16]YUKSELEN M A,GREGORY J.Breakage and reformation of alum flocs[J].Environmental Engineering Science,2002,19(4):229-236.
[17]YU W Z,LI G B,XU Y P,et al.Breakage and regrowth of flocs formed by alum and PACl[J].Powder Technology,2009,189:439-443.
[18]YU W Z,GREGORY J,CAMPOS L C.Breakage and re-growth of flocs formed by charge neutralization using alum and polyDADMAC [J].Water Research,2010,44:3959-3965.
[19]WANG D S,WU R B,JIANG Y Z,et al.Characterization of floc structure and strength:role of changing shear rates under various coagulation mechanisms[J].Colloids and Surfaces A:Physicochemical and Engineering Aspects,2011,379:35-42.
[20]RONGH Y, GAOBY, DONGM, etal.Characterization of size,strength and structure of aluminum-polymer dual-coagulant flocs under different pH and hydraulic conditions[J].Journal of Hazardous Materials,2013,252/253:330-337.
[21]VAHEDIA,GORCZYCA B.Application offractal dimensions to study the structure of flocs formed in lime softening process[J].Water Research,2011,45:545-556.
[22]YU W Z,GREGORY J,CAMPOS L.Breakage and regrowth of Al-Humic flocs-effect of additional coagulant dosage [J].Environmental Science and Technology,2010,44:6371-6376.
[23]ZHAO Y X,GAO B Y,SHON H K,et al.The effect of second coagulant dose on the regrowth of flocs formed by charge neutralization and sweep coagulation using titanium tetrachloride (TiCl4) [J].Journalof Hazardous Materials,2011,198:70-77.
[24]XOAO F,ZHANG X R,LEE C.Is electrophoretic mobility determination meaningful for aluminum(III)coagulation of kaolinite suspension[J].Journal of Colloid and Interface Science,2008,327:348-353.
[25]YU W Z,GREGORY J,CAMPOS L.Breakage and regrowth of flocs:effect of additional doses of coagulant species[J].Water Research,2011,45:6718-6724.
