含氮化合物對FCC催化劑的中毒機理及其應對措施
2013-11-05 05:34:58沈本賢陳小博楊朝合山紅紅
石油化工 2013年4期
沈本賢,陳小博,,王 勁,楊朝合,山紅紅
(1. 華東理工大學 化工學院,上海 200237;2. 中國石油大學(華東) 重質油國家重點實驗室,山東 青島 266580)
近幾年來,隨著FCC裝置摻煉渣油、焦化蠟油(CGO)以及頁巖油等高含氮原料比例的不斷增加,如何抑制含氮化合物(尤其是堿性含氮化合物)對FCC催化劑的毒害作用、提高FCC裝置有效加工劣質重油的能力,已成為煉廠急需解決的難題之一[1-4]。目前,抑制或消除含氮化合物對FCC過程不利影響的措施主要包括兩個方面:一是針對FCC原料,進行加氫脫氮[5]、絡合脫氮[6-7]、吸附脫氮[8]、溶劑抽提脫氮[9]和酸中和脫氮[10]等原料預處理過程;二是采用FCC抗氮工藝、使用抗氮催化劑或助劑。在這些措施中,原料加氫脫氮被認為是最有效的方法,但受加氫裝置投資成本和操作成本高的限制,尤其在我國氫源和加氫能力十分有限的背景下,FCC原料加氫脫氮工藝在工業中的大規模應用受到嚴重影響。其他原料脫氮工藝則存在脫氮率低、油收率低、脫氮劑回收困難和脫氮成本高等缺點,鮮有工業應用報道。因此,通過對工藝過程的改進或使用抗氮催化劑及助劑,提高FCC裝置加工高含氮原料的能力,在目前被認為是一種比較經濟有效的途徑[11]。關于含氮化合物對FCC催化劑的中毒作用機理及其應對措施的研究引起了研究者和煉油工作者的極大關注。
本文介紹了含氮化合物對FCC催化劑中毒作用機理的研究現狀,概述了各種抗氮催化劑和抗氮工藝的研究進展,列舉了抑制含氮化合物毒害FCC催化劑的應對措施,展望了提高FCC裝置加工高含氮原料的發展方向。
1 FCC催化劑的氮中毒機理
FCC原料中的氮原子多數存在于大分子中,還常常與其他雜原子(如硫、氧等)和金屬離子共存,所以含氮化合物的結構和組成極其復雜,物理化學性質也差別很大,這給研究含氮化合物對FCC催化劑的中毒作用機理帶來了巨大挑戰。因此,盡管早在無定形硅鋁催化劑時代,研究者就對含氮化合物會嚴重影響FCC催化劑的活性進行了報道,但幾十年過去了,對含氮化合物對FCC催化劑的中毒作用機理仍沒有統一的認識,不同的研究人員根據不同的研究體系,提出了不同的中毒作用機理。
1.1 酸堿中和理論
FCC過程中烴類的大部分反應都是在固體酸催化劑的作用下,按碳正離子機理進行。該過程的催化活性中心包括B酸和L酸中心,前者有能力向堿貢獻一個質子(H+),而后者則能從堿中接受一個未成對的電子。含氮化合物因具有堿性特征,很容易與催化劑上的酸性中心發生相互作用,造成催化劑活性下降,酸堿中和理論正是基于此提出的。該理論認為含氮化合物,主要是堿性含氮化合物與催化劑B酸中心提供的H+結合,使B酸中心數目減少;或者是含氮化合物向催化劑表面配位不飽和的Al原子或Si原子(L酸中心)提供未成鍵的孤對電子,從而引起催化劑失活[11-12],其中毒過程如圖1和圖2所示。根據這一理論,含氮化合物對催化劑的毒害作用強弱應該與其堿性大小有很大的關系,但研究結果[13-14]卻表明,含氮化合物毒害作用的強弱與其堿性大小并沒有一致性,而是與含氮化合物在氣相狀態下的質子親和力有較好的對應關系。這可能是由于含氮化合物堿性大小的測定是在常溫下進行的,與FCC高溫條件下的氣、液、固多相體系相差甚遠,而含氮化合物在氣相狀態下的質子親和力則能更好地反映含氮化合物在FCC體系中與B酸或L酸中心的結合能力。
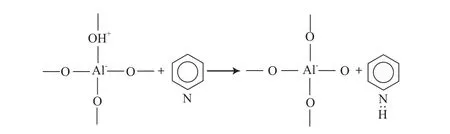
圖1 催化劑B酸中心氮中毒示意Fig.1 Diagram of the nitrogen poisoning of the B acid sites on a FCC catalyst.
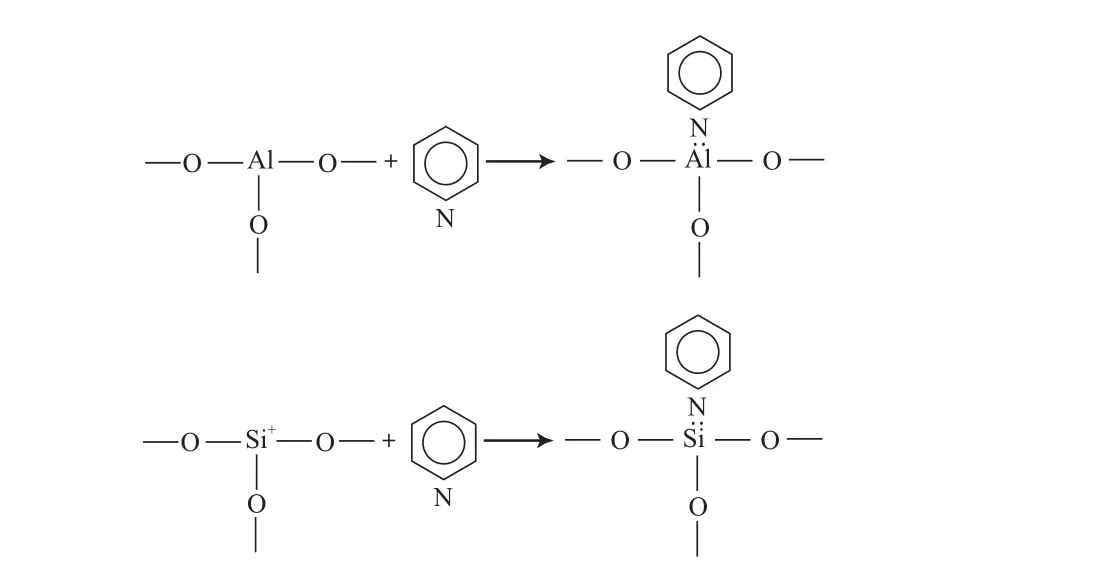
圖2 催化劑L酸中心氮中毒示意Fig.2 Diagram of the nitrogen poisoning of the L acid sites on a FCC catalyst.
1.2 競爭吸附理論
含氮化合物一般都以多環芳烴的形式存在,且含氮雜環具有很強的吸附和絡合能力,與多環芳烴、單環芳烴、環烷烴、烯烴和烷烴等相比,更易在催化劑表面酸性中心上發生吸附。因此競爭吸附理論認為,含氮化合物會優先吸附在催化劑的酸性中心上,但同時由于含氮雜環很難發生裂化反應,極易在吸附活性位上產生焦炭前身物,繼而縮合生成焦炭,覆蓋催化劑的活性中心或堵塞分子篩的孔道,造成催化劑活性下降。Barth等[15]利用紅外光譜、核磁共振和基質輔助激光解吸電離飛行時間質譜對結焦催化劑上的含氮化合物進行研究時發現,焦炭中的含氮化合物大多數是含有六環以上的甲基喹啉類和甲基四氫喹啉類的衍生物,其質荷比在350~850之間,主要分布在催化劑的介孔和大孔中,說明含氮化合物在催化劑上吸附以后,主要發生縮合生焦反應。Caeiro等[16-17]認為,FCC催化劑在反應過程的初期,活性和選擇性急劇降低的主要原因是USY分子篩超籠中的酸性中心被焦炭所覆蓋,而這恰恰是由于堿性含氮化合物優先吸附在B酸中心上并縮合生焦導致的。Li等[18]和劉銀東等[19-20]的研究結果也表明,含氮化合物會不可逆地優先吸附在催化劑的酸性中心上,導致催化劑活性中心數目減少或作為焦炭前身物在催化劑表面縮合生焦。
1.3 誘導效應和空間位阻效應
Corma等[21]根據吡啶、2,6-二甲基吡啶和喹啉對正庚烷在USY分子篩催化劑上裂化性能的影響規律,提出了堿性含氮化合物對活性中心的中毒作用是通過誘導效應進行的。他們認為含氮化合物分子,尤其是帶有多個側鏈的含氮化合物分子,在酸中心上吸附后,與其直接作用的B酸中心電荷密度大幅降低,同時含氮化合物分子上剩余電荷通過誘導效應向鄰近的B酸中心轉移,改變了其原有的電荷密度,使其不足以催化碳正離子反應,即一個含氮化合物分子可能會造成多個活性中心失去活性。Caeiro等[22-23]通過FTIR分析,在解釋2,6-二甲基吡啶對催化劑的中毒作用強于3-甲基吡啶和喹啉時認為,分子中兩個甲基的空間位阻效應,導致2,6-二甲基吡啶只能吸附在B酸中心上,而3-甲基吡啶和喹啉分子則可同時吸附于B酸和L酸中心上,由于在FCC反應初期,主要是B酸中心起作用,因此2,6-二甲基吡啶對催化劑的毒害作用強于另外兩種堿性含氮化合物,即L酸中心分擔了一部分3-甲基吡啶和喹啉對B酸中心的毒害作用。根據誘導效應和空間位阻效應,含氮化合物對催化劑的中毒作用的強弱與其分子大小和結構有很大的關系。
綜上所述,酸堿中和理論與含氮化合物在氣相條件下的堿性(即質子親和力)有很大的關系;競爭吸附理論與含氮化合物的吸附和絡合能力,以及吸附后是否會在催化劑活性中心上參與生焦反應有關;而誘導效應和空間位阻效應則與含氮化合物的分子大小和分子結構(帶支鏈情況),以及催化劑活性組分的孔結構等密切相關。因此,由于原料中含氮化合物的復雜性以及FCC催化劑活性組分的不同,含氮化合物對FCC催化劑的毒害也呈現出不同的作用機理,很難用某一個指標來預測含氮化合物的中毒能力,只有從含氮化合物的堿性、分子結構、分子大小、吸附和絡合能力,催化劑的活性組分以及含氮化合物在原料中的濃度大小等多方面綜合考慮,才能真實反映含氮化合物對FCC催化劑的影響。
2 應對措施
目前,針對含氮化合物對FCC催化劑的中毒作用,所采取的應對措施主要包括兩個方面:一是開發抗氮催化劑,通過提高小孔分子篩的應用比例以限制大分子含氮化合物的吸附效應;優化分子篩酸量及酸類型,提高目的產品的選擇性;同時配合活性基質,提高重油的轉化能力。二是通過改變操作條件和開發新工藝,提高FCC裝置處理高含氮原料的能力。
2.1 FCC抗氮催化劑
2.1.1 增加活性中心數量
根據催化劑的氮中毒機理,含氮化合物的毒害作用無論是因為酸堿中和引起活性中心數目減少,還是由于競爭吸附、誘導效應、空間位阻效應導致活性中心被覆蓋,歸根到底都會造成用于催化烴類分子裂化的活性中心數量的減少,因此增加催化劑活性中心的數量,能減輕含氮化合物對烴類裂化反應的影響。Scherzer等[24]研究認為,催化劑中分子篩含量高對提高抗氮性能尤為重要。Young等[25]也認為在目前的FCC催化體系中,增加活性中心數量是解決催化劑氮中毒最好的方法。通常情況下,增加催化劑中分子篩的含量,就相當于增加了活性中心的數量,堿性含氮化合物含量較低時,催化劑的抗氮能力隨分子篩含量的增加而增加;但當堿性含氮化合物含量較高時,分子篩含量高的催化劑活性衰減反而更為劇烈。這可能是由于過多的分子篩使含氮化合物在催化劑表面的生焦反應加速,焦炭覆蓋了更多的活性中心或堵塞孔道所致。因此,FCC催化劑的分子篩含量必須根據具體情況,在合適的范圍內進行調節[26]。
2.1.2 使用活性基質
利用活性基質上的酸性中心吸附含氮化合物,可以消除或抑制含氮化合物對催化劑分子篩的毒害作用,從而提高FCC催化劑的抗氮性能。Scherzer等[24]認為活性基質的引入使重油轉化率和汽油的選擇性均有所提高,表明活性基質對含氮化合物的吸附效應明顯,在一定程度上保護了分子篩的活性。Corma等[27]對采用惰性載體和活性載體的USY分子篩催化劑的抗氮性能進行了對比研究,也得出了類似的結論。因此,提高催化劑基質的活性是提高催化劑抗氮性能的有效途徑之一,但由于基質酸性的增加促進了重油大分子在基質表面的裂化反應和縮合生焦反應,從而造成焦炭產率增加。
2.1.3 分子篩改性
分子篩的硅鋁比、酸密度和孔分布等,對催化劑的抗氮性能也會產生影響。低硅鋁比、高酸中心密度和合適的孔分布對提高催化劑的抗氮性能更為有利,因此,許多研究者通過對分子篩進行改性,來提高催化劑的抗氮性能。USY分子篩是FCC領域應用最為廣泛的一種分子篩,可通過對USY分子篩的酸密度和酸類型進行調變來提高其抗氮性能。目前,用于USY分子篩抗氮改性的元素主要有P,La,Zn,Mn,Cr以及稀土和堿土金屬[28-30]。袁啟民等[30]在小型固定床反應器中系統考察了La改性后USY分子篩的抗氮性能,研究結果表明,La的引入可使催化劑的總酸量明顯增加,抗氮性能有所提高,但焦炭產率增加也較為明顯。稀土改性可改善USY分子篩的抗氮性能,但要解決焦炭產率增加的問題[26]。
在開發新型抗氮催化劑時,應該選擇具有合適孔徑和孔分布的分子篩,以阻止大分子含氮化合物進入孔道內部,然后再通過元素改性調變分子篩的酸密度和酸類型,同時選擇活性基質,有效分散含氮化合物在催化劑上的吸附,進而保護分子篩活性,并適當提高分子篩含量,改善重油轉化率和產物分布。但在提高催化劑抗氮性能的同時,必須兼顧催化劑焦炭產率增加的矛盾。近年來,國內已開發出一批適合摻煉CGO的稀土HY型催化劑,如中國石化長嶺催化劑廠生產的CC-14和CC-15型催化劑,中國石化齊魯催化劑廠生產的RHZ-200催化劑及其改進型RHZ-300催化劑,以及中國石油蘭州催化劑廠生產的LCS-7和LC-8型催化劑等[11],都是以稀土改性后的HY分子篩為活性組分。工業應用結果表明,與常規催化劑相比,這些催化劑具有較高的重油轉化能力和抗氮性能,可使FCC裝置的CGO摻煉比提高10%~15%。
2.2 FCC抗氮工藝
2.2.1 優化操作條件
反應溫度越高越有利于脫附,因此升高反應溫度會減弱含氮化合物在催化劑上的吸附,抑制含氮化合物的毒害作用,從而使產品分布得到改善。在實際生產中,為了提高FCC裝置摻煉CGO的能力,往往采用升高反應溫度的措施,如中國石化長嶺煉油廠將反應溫度由490~495 ℃升高到500~505 ℃,CGO摻煉比由原來的21.0%增至25.4%[12]。此外,提高劑油比以及增大原料油分子與催化劑活性中心的接觸機會,也有利于高含氮原料的裂化。
2.2.2 催化裂化吸附轉化工藝
根據CGO與常規催化原料性質的不同,中國石化石油化工科學研究院開發了CGO催化裂化吸附轉化(DNCC)工藝[31]。該工藝的特點是將易于裂化的優質催化原料(通常為減壓蠟油)與CGO分層進料,優質催化原料從提升管的底部進入,先與高活性的再生劑接觸并發生反應,CGO從提升管的中部或后半段進入,與已結焦部分失活的半待生催化劑接觸,利用相對低溫的半待生催化劑吸附CGO中較難轉化的稠環芳烴和含氮化合物,使CGO部分轉化并達到精制的目的。同時CGO可以起到終止劑的作用,有利于提高優質催化原料的劑油比和反應溫度,改善其產物分布。精制后的CGO再以回煉油的方式返回提升管,繼續進行反應,其原理示意如圖3所示。雖然DNCC工藝可使CGO在相對緩和的條件下脫除其中的含氮化合物,但不難發現,該工藝是靠犧牲提升管后半段的裂化作用以達到精制CGO的目的,這樣不可避免地會造成回煉比增大、能耗以及操作費用的增加[12]。
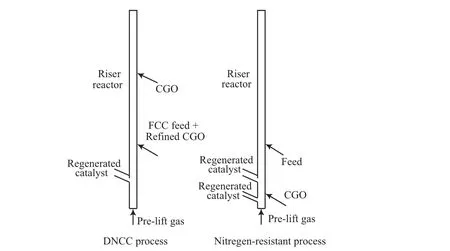
圖3 DNCC工藝和Chevron公司抗氮裂化工藝的原理Fig.3 Diagrams of DNCC process and nitrogen-resistant process of Chevron.
2.2.3 Chevron公司的抗氮裂化工藝
Chevron公司開發的抗氮裂化工藝的原理(如圖3所示)與DNCC工藝類似,同樣是利用催化劑預先與CGO發生吸附反應,以降低含氮化合物對主反應的影響。不同之處在于,該工藝是利用原料在進入提升管主反應區之前,使其與少量的再生催化劑接觸,以吸附原料中的部分堿性含氮化合物,靠“犧牲”這部分催化劑的活性來減輕堿性含氮化合物對主反應的影響,從而達到提高原料轉化率的目的[11]。實驗結果表明,在預接觸區劑油比為1時,最高脫氮率可達31%,轉化率提高了11百分點,而干氣和焦炭產率并沒有明顯的變化。
2.2.4 CGO直接催化裂解技術
利用大分子含氮化合物很難進入到ZSM-5分子篩的孔道中,從而對以ZSM-5分子篩為活性組分的多產丙烯催化劑中毒作用相對較小的特點,結合兩段提升管FCC技術的優勢,并采用焦化蠟油與焦化石腦油組合進料技術,中國石油大學(華東)開發了CGO直接催化裂解技術,在促進CGO有效轉化的同時,提高了焦化石腦油的辛烷值,并獲得了較高的丙烯收率。該技術的300 kt/a工業示范裝置已于2011年9月4日在山東恒源石油化工股份有限公司順利投產,在CGO與焦化石腦油按質量比2∶1進料的情況下,總液體質量收率近90%,液化氣質量收率20%左右,液化氣中丙烯的含量達47%(φ)以上,汽油的研究法辛烷值約為90,為高含氮原料的直接加工利用開辟了一條新的途徑[32]。
3 結語
隨著FCC裝置摻煉渣油和CGO等高含氮原料比例的不斷增加,催化劑受氮中毒污染的問題將更加突出。通過FCC原料預處理、采用抗氮催化劑或助劑以及調整FCC裝置的操作條件和工藝路線,都可在一定程度上緩解含氮化合物對催化劑的毒害作用,提高FCC裝置的重油轉化能力,改善其產物分布。雖然原料加氫脫氮被認為是最有效、資源利用率最高的方法,但采用抗氮催化劑或助劑是目前最經濟有效的措施。因此,在深入認識FCC催化劑氮中毒機理的基礎上,對FCC催化劑進行抗氮功能設計,通過選用合適的活性組分或對分子篩進行改性,調變分子篩的酸密度和孔分布,同時選擇活性基質,并采用適當提高分子篩含量等方法,開發具有良好抗氮性能的催化劑,以滿足加工重油的需要,仍是當前煉油行業的發展趨勢之一。
[1] 闕國和. 石油組成與轉化化學[M]. 東營:中國石油大學出版社,2008:100 - 122.
[2] Wang Gang,Li Zekun,Huang He,et al. Synergistic Process for Coker Gas Oil and Heavy Cycle Oil Conversion for Maximum Light Production[J]. Ind Eng Chem Res,2010,49(22):11260 - 11268.
[3] Cerqueira H S,Caeiro G,Costa L,et al. Deactivation of FCC Catalysts[J]. J Mol Catal A:Chem,2008,292(1/2):1 - 13.
[4] 蔡開鵬. 加工焦化蠟油對催化裂化裝置的影響及技術對策[J]. 石油學報:石油加工,2010,26(增刊):59 - 65.
[5] Wiwel P,Hinnemann B,Hidalgo-Vivas A,et al. Characterization and Identification of the Most Refractory Nitrogen Compounds in Hydroprocessed Vacuum Gas Oil[J]. Ind Eng Chem Res,2010,49(7):3184 - 3193.
[6] 馬麗娜,馬守濤,劉麗瑩,等. 焦化蠟油絡合脫氮-催化裂化組合工藝研究[J]. 石油與天然氣化工,2011,40(6):571 - 573.
[7] 郭立艷,萬書寶,趙光輝,等. 焦化蠟油絡合脫氮用作催化裂化摻煉進料的研究[J]. 石油煉制與化工,2008,39(10):18 - 21.
[8] 馬駿,隋新,王海彥. 微波處理吸附劑脫除堿性氮化物的研究[J]. 石油化工高等學校學報,2004,17(2):9 - 11.
[9] 黃新龍,曲賀欣,王更新,等. 延遲焦化-溶劑精制及其組合工藝研究[J]. 煉油設計,2001,31(3):36 - 39.
[10] 凌昊,沈本賢,高玉延,等. 甲酸絡合萃取脫除焦化蠟油中堿性氮化物的研究[J]. 石油煉制與化工,2003,34(8):28 - 31.
[11] 袁起民,龍軍,謝朝鋼,等. 高氮原料的催化裂化研究進展[J]. 化工進展,2008,27(2):1929 - 1936.
[12] 袁起民. 焦化蠟油催化裂化轉化應用基礎研究[D]. 青島:中國石油大學(華東),2007.
[13] Fu Chia Min,Schaffer A M. Effect of Nitrogen Compounds on Cracking Catalysts[J]. Ind Eng Chem Prod Res Dev,1985,24(1):68 - 75.
[14] Ho Teh Chung,Katritzky A R,Cato S J. Effect of Nitrogen Compounds on Cracking Catalysts[J]. Ind Eng Chem Res,1992,31(7):1589 - 1597.
[15] Barth J O,Jentys A,Lercher J A. On the Nature of Nitrogen-Containing Carbonaceous Deposits on Coked Fluid Catalytic Cracking Catalysts[J]. Ind Eng Chem Res,2004,43(10):2368 - 2375.
[16] Caeiro G,Magnoux P,Lopes J M,et al. Deactivating Effect of Quinoline During the Methylcyclohexane Transformation over H-USY Zeolite[J]. Appl Catal,A,2005,292(9):189 - 199.
[17] Caeiro G,Costa A F,Cerqueira H S,et al. Nitrogen Poisoning Effect on the Catalytic Cracking of Gasoil[J]. Appl Catal,A,2007,320(3):8 - 15.
[18] Li Zekun,Wang Gang,Shi Quan,et al. Retardation Effect of Basic Nitrogen Compounds on Hydrocarbons Catalytic Cracking in Coker Gas Oil and Their Structural Identification[J]. Ind Eng Chem Res,2011,50(7):4123 - 4132.
[19] 劉銀東,李澤坤,王剛,等. 競爭吸附對催化裂化反應過程的影響[J]. 化工學報,2008,59(11):2794 - 2799.
[20] 劉銀東,王剛,李澤坤,等. 焦化蠟油催化裂化反應過程生焦特性[J]. 石油學報:石油加工,2009,25(1):7 - 13.
[21] Corma A,Fornés V,Montón J B,et al. Catalytic Cracking of Alkanes on Large Pore,High SiO2/Al2O3Zeolites in the Presence of Basic Nitrogen Compounds:Influence of Catalyst Structure and Composition in the Activity and Selectivity[J].Ind Eng Chem Res,1987,26(5):882 - 886.
[22] Caeiro G,Magnoux P,Lopes J M,et al. Deactivating Effect of Coke and Basic Nitrogen Compounds During the Methylcyclohexane Transformation over H-MFI Zeolite[J]. Chem Eng J,2006,120(1/2):43 - 54.
[23] Caeiro G,Lopes J M,Magnoux P,et al. A FT-IR Study of Deactivation Phenomena During Methylcyclohexane Transformation on H-USY Zeolites:Nitrogen Poisoning,Coke Formation,and Acidity-Activity Correlations[J]. J Catal,2007,249(2):234 - 243.
[24] Scherzer J,McArthur D P. Tests Show Effects of Nitrogen Compounds on Commercial Fluid Catalytic Cracking Catalysts[J]. Oil Gas J,1986,84(43):76 - 82.
[25] Young G W. Fluid Catalytic Cracker Catalyst Design for Nitrogen Tolerance[J]. J Phys Chem,1986,90(20):4894 - 4900.
[26] 孫金鵬. 焦化蠟油兩段提升管催化裂解與焦化石腦油改質應用基礎研究[D]. 青島:中國石油大學(華東),2011.
[27] Corma A,Mocholi F A. New Silica-Alumina-Magnesia FCC Active Matrix and Its Possibilities as a Basic Nitrogen Passivating Compound[J]. Appl Catal,A,1992,84(5):31 - 46.
[28] 孫書紅,龐新梅,鄭淑琴,等. 稀土超穩Y型分子篩催化裂化催化劑的研究[J]. 石油煉制與化工,2001,32(6):25 - 28.
[29] 朱華元,何鳴元,宋家慶,等. 堿土金屬分子篩對FCC催化劑催化性能的影響[J]. 石油學報:石油加工,2001,17(6):6 - 9.
[30] 袁啟民,張兆濤,李春義,等. 鑭改性對催化裂化催化劑物化性能及抗氮性能的影響[J]. 中國石油大學學報:自然科學版,2008,32(2):127 - 131.
[31] 張瑞馳,施文元. 催化裂化吸附轉化加工焦化蠟油工藝的研制與開發[J]. 石油煉制與化工,1998,29(11):22 - 27.
[32] 王建高,劉積舜. 我國焦化蠟油直接催化裂解技術出新[N].科技日報,2012-01-16(1).
猜你喜歡
山東冶金(2019年6期)2020-01-06 07:45:54
世界農藥(2019年2期)2019-07-13 05:55:12
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
銅業工程(2015年4期)2015-12-29 02:48:39
新疆鋼鐵(2015年3期)2015-11-08 01:59:52
應用化工(2014年3期)2014-08-16 13:23:50
