ADMA/DDAH 通路在腦缺血再灌注致肺微血管內皮細胞損傷中的作用
2013-03-11 08:19:50吳云虎王殿華
中風與神經疾病雜志 2013年10期
吳云虎,王殿華
ADMA/DDAH 通路在腦缺血-再灌注機制中研究較多,但在腦缺血-再灌注致肺損傷中對PMVECs通透性的作用機制仍未見有報道。本研究探討ADMA/DDAH 通路調控PKCα 和MCK 蛋白表達對PMVEC 的影響,進一步揭示ADMA/DDAH 通路在腦I/R 致ALI 中的機制。
1 材料與方法
1.1 材料 實驗動物:雌雄各半SD 大鼠36只,每體重200~260g,購自廣西醫科大學實驗動物中心。主要試劑:ADMA 標準品購自Sigma 公司;MTT 試劑購自Biomol 公司;逆轉錄試劑盒購自美國Promega 公司;2×Taq PCR MasterMix 購自北京天根生化科技有限公司;RIPA 裂解液、PKC 和MLCK 蛋白濃度測定試劑盒購自江蘇碧云天公司;RT-PCR試劑盒,批號:9440-94A。
1.2 方法
1.2.1 大鼠腦缺血再灌注模型的建立 參照Zea-Longa 改良線栓法制作大腦中動脈缺血模型(MCAO);參照Zea-Longa 大鼠神經功能缺損評分標準對MCAO 進行評分。
1.2.2 腦缺血再灌注損傷大鼠血清的采集各組大鼠按設定處死時間,以氯胺酮麻醉后頸動脈置管,取1.0ml 血置于檸檬酸抗凝管搖勻,3000 轉/min 離心,留取上清。分為滅活血清組和非滅活血清組處理:選用與血清瓶同規格的對照瓶一個,對照瓶內放入與血清等體積的水,調節水浴箱溫度控制鈕,使溫度保持在56℃,定時30min 進行血清滅活,滅活后-20℃~-70℃保存。
1.2.3 大鼠PMVECs 的培養 取體健康雄性Wistar 大鼠1 只,麻醉固定后,取出肺臟至DMEM 培養液中,切割至約1mm3大小的組織塊,均勻放置在25ml 培養瓶中,靜置培養60h 后去除組織塊,于80%~90%細胞單層匯合時用0.25%胰蛋白酶消化后1∶2 傳代。
1.2.4 細胞傳代 當EC 對數生長至細胞密度達80%左右傳代,用D-Hank’s:液漂洗3 次,加0.25%胰養蛋白酶1ml 于瓶中,使細胞充分接觸消化液,用吸管吹打細胞懸液,將此瓶細胞懸液分成二份加入新瓶中,放入培養箱中繼續培養。以下實驗采用2~5 代細胞進行。
1.2.5 實驗分組 將大鼠PMVECs 放在100ml 培養瓶內,待80%單層匯合時進行實驗,隨機分為5 組:正常細胞組、正常滅活血清組、正常非滅活血清組、模型滅活血清組、模型非滅活血清組、ADMA 干預組和DDAH 抑制劑干預組。以上各組在培養箱中孵育4h,檢測各項指標。
1.2.6 RT-PCR 法檢測PKC 和MLCK mRNA的表達 按Trizol 法提取總mRAN,按照一步法RTPCR 試劑盒說明書操作,獲得目的基因DNA 片段。引物序列:上游引物 5’-TGTCCAAAACCTACCCCACCATAT-3’,下游引物5’-CCCTTCTTACATCAGCCCTACTG-3’,擴增片段為73bp 大小(引物合成由日本TaKaRa 公司合成)。瓊脂糖凝膠電泳檢測擴增產物,Gel-Doc 紫外凝膠成像分析系統計算各條帶的灰度值。
1.2.7 Western blot 檢測細胞PKC 和MLCK 的表達 在細胞培養瓶中在37℃,5%CO2條件下培養,當細胞密度近80%時,oxLDL 刺激6h 后加不同試劑,培養40min 檢測信號蛋白表達,12h 后檢測PKCα 和MLCK 表達,用PBS(pH7.4)洗3 遍,加入0.5ml RIPA buffer,用細胞刮子將細胞刮下來,放到1.5ml 的EP 管中,冰上放置30min;在超速冷凍離心機中14000rpm 離心20min,保留上清;用Lorry 法對不同的細胞裂解產物定量其總蛋白濃度;按1∶1的比例加入2×蛋白上樣緩沖液,沸水煮5min;SDSPAGE 電泳125g/L 使用聚丙烯酰胺凝膠。封閉液在室溫下封閉1h,加入抗myc 抗體4℃過夜。加入辣根過氧化物酶標記的抗兔IgG 抗體形成免疫復合物,DAB 顯色后攝像。
1.2.8 MTT 法細胞增殖/抑制活性測定 取經不同濃度處理24h 的細胞孔,加入體積分數為0.05g/L MTT 10μl/孔繼續培養4h,取板,吸盡上清,各孔加DMSO 100μl,室溫避光震蕩10min,讀取A492 值(OD)/孔。依據實驗分組,對各干預組測定3 個平行孔A490 值(OD),細胞增殖率(%)=[實驗組490A 值(OD)/正常對照組490A 值(OD)]×100%,其結果均以增殖指數>100% 表示細胞刺激增殖作用。同樣方法測定A570 值(OD)。
2 結果
2.1 PMVEC 中PKCα、MLCK 蛋白的表達 實驗結果顯示:與其余3 組比較,模型滅活血清組、模型非滅活血清組和ADMA 組PMVEC 中PKCα 和MLCK 蛋白水平明顯升高(P<0.05),差異有統計學意義,其余3 組之間PKCα 和MLCK 蛋白水平比較差異無統計學意義(P>0.05)(見表1、圖1)。
2.2 PMVEC 中PKCα、MLCK mRNA 的表達實驗結果顯示:與其余3 組比較,模型滅活血清組、模型非滅活血清組和ADMA 組PMVEC 中PKCα、MLCK mRNA 表達明顯升高(P<0.05),差異有統計學意義,其余3 組之間PKCα 和MLCK 蛋白水平比較差異無統計學意義(P>0.05)(見表2、圖2)。
2.3 PMVEC 細胞增殖活性 與其余4 組OD值(λ=490 波長)比較,模型滅活血清組和ADMA組有統計學意義(P<0.05),細胞代謝活力顯著降低;與ADMA 組相比較,DDAH 組OD 值顯著增高,細胞代謝活力顯著增強(P<0.05),差異有統計學意義(見表3)。
表1 PMVEC 中PKCα、MLCK 蛋白表達灰度值(±s)
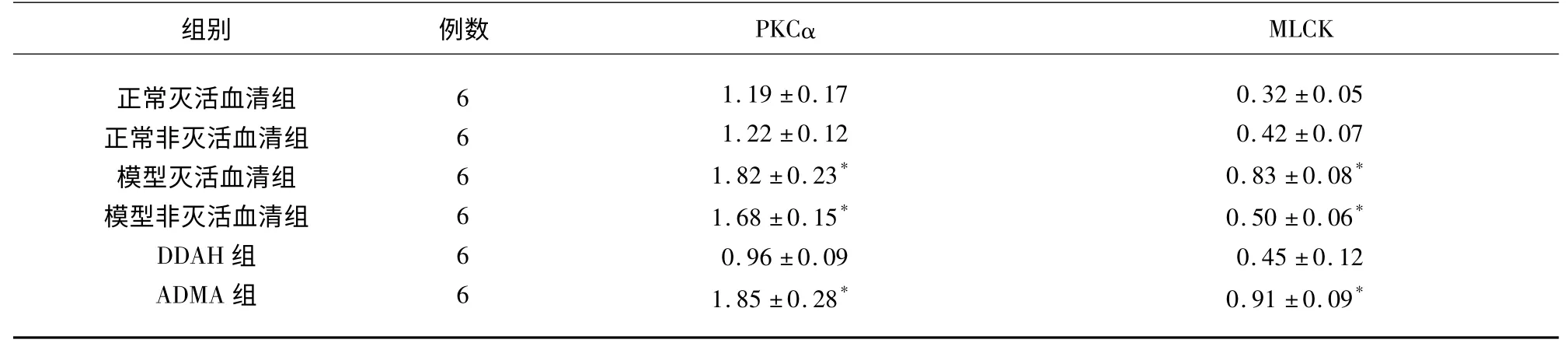
表1 PMVEC 中PKCα、MLCK 蛋白表達灰度值(±s)
與正常滅活血清組、正常非滅活血清組和DDAH 干預組比較* P<0.05
表2 PMVEC 中PKC、MLCK mRNA 表達灰度值(±s)
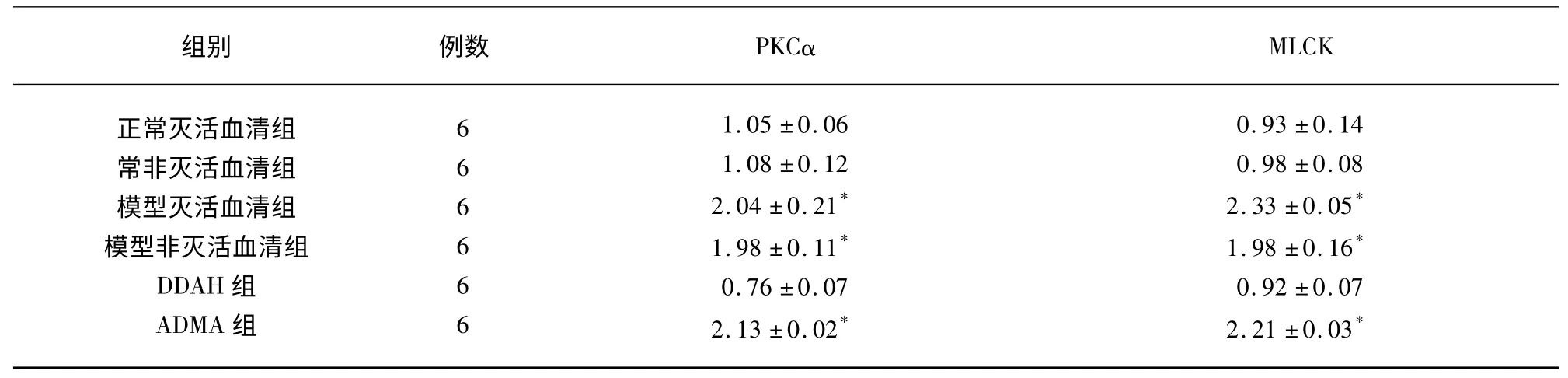
表2 PMVEC 中PKC、MLCK mRNA 表達灰度值(±s)
與正常滅活血清組、正常非滅活血清組和DDAH 干預組比較* P<0.05
表3 各組的細胞增殖(±s)
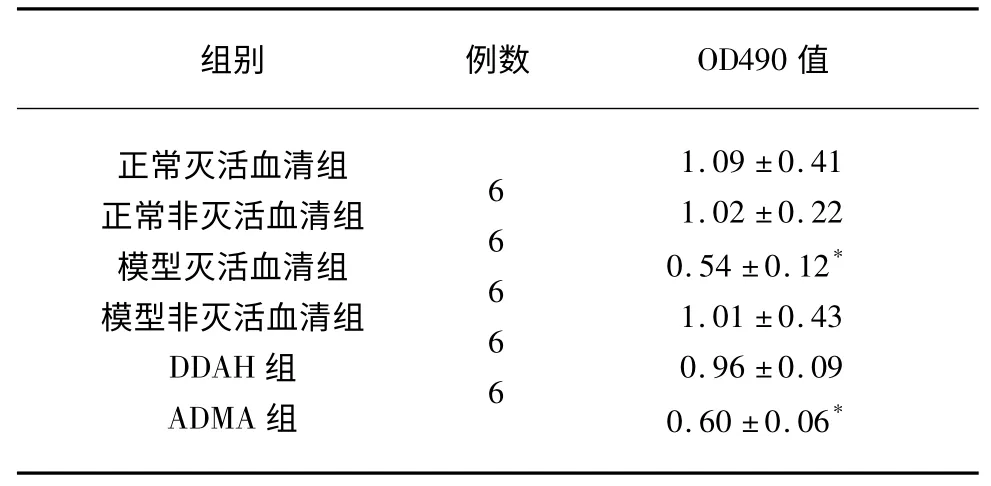
表3 各組的細胞增殖(±s)
與其余4 組比較* P<0.01
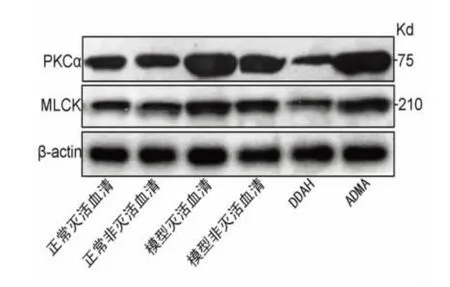
圖1 腦I/R 損傷致ALI 細胞模型中PMVEC 中PKCα、MLCK 蛋白表達。與正常滅活血清組、正常非滅活血清組和DDAH 干預組比較* P<0.05

圖2 腦I/R 損傷致ALI 細胞模型中PMVEC 中PKCα、MLCK mRNA 表達。與正常滅活血清組、正常非滅活血清組和DDAH 干預組比較* P<0.05
3 討論
新近研究證實[1,2]ADMA/DDAH 通路在體內多個器官的細胞中存在,該通路參與多種生理和病理過程,在許多疾病的發生發展中起重要作用。
ADMA 在蛋白精氨酸甲基轉移酶(protein arginine methytransferases,PRMT)作用下,以S-腺苷甲硫氨酸(S-adenosylme-thionine,SAM)為甲基供體,使各種多肽中的L-精氨酸殘基甲基化,甲基化的蛋白質水解以后產生。ADMA 的主要降解及排泄系統是在二甲基精氨酸二甲氨水解酶(dimethylargininedimethy laminohydrolase,DDAH)作用下分解為瓜氨酸和二甲胺經腎臟排出,所以PRMT 及DDAH 控制著ADMA 在體內的濃度,ADMA 的代謝途徑即ADMA/DDAH 通路[3,4]。研究發現,有多種細胞能生成ADMA,包括血管內皮細胞,健康人血漿中ADMA 的濃度為(1.15±0.13)μmol/L,內皮細胞中的精確濃度尚未得知,但細胞內ADMA 的濃度大約是培養液中的8~12 倍[5,6]。
ADMA 是內源性NOS 抑制劑。大量的研究證實ADMA 能競爭性抑制NOS,阻止NO 合成,增加氧自由基的生成和引發血管炎癥。近年來的研究發現ADMA 聚集是內皮功能不全的一個重要原因,其增高預示心血管疾病的患病風險和死亡率增高[7]。DDAH 的表達增強可以起到抑制ADMA 生成的作用,使用維甲酸上調DDAH 表達可以使血管內皮細胞ADMA 表達減少,含量下降,NO 合成增加。因此,DDAH 是一種比較理想的血管保護因子,ADMA/DDAH 通路可能在氧化應激反應和內皮保護作用之間產生有意義的影響,合理調整DDAH 含量,增強其表達,抑制ADMA 含量的增加,降低其表達,可以對缺血再灌注損傷起到很好的保護作用[8,9]。
PKC 的持續高表達和異常激活是導致肺微血管內皮細胞形態學變化的最主要原因。PKC 激活后對肌動蛋白肌絲相關蛋白進行磷酸化,并能夠使肌球蛋白ATP 酶活性增加,從而使肌動蛋白和肌球蛋白相互結合維持持續的肌絲的收縮[10];PKCα 亦可參與促進絲裂素活化蛋白激酶(MAPK)的激活過程,而MAPK 的激活在維持平滑肌的持續收縮和膠原纖維增生中起重要作用[11];另外,PKCα 還可通過調節蛋白酪氨酸激酶(PTK)Rho 蛋白的磷酸化來調節肌絲的收縮[12];MLCK 介導了血管內皮細胞的損害,導致通透性增加,引起血管內皮細胞間隙的形成和屏障功能障礙[13]。
經ADMA 干預后,MLCK 的蛋白表達進一步增強,提示ADMA 可以增加內皮細胞通透性,導致MLCK 活性不斷升高,MLCK 催化MLC 磷酸化程度加大,導致EC 的向心性收縮增強,細胞間隙形成程度加大,從而在一定程度上加重了氧化應激反應和炎癥反應的發展。經過DDAH 干預后,血管內皮細胞的滲透程度有所下降,細胞的通透性得到修復,內膜增厚,此修復的結果使得MLCK 的蛋白表達和蛋白含量明顯下降,并能降低MLCK 的轉錄,阻止缺血再灌注肺損傷的進一步發展,在一定程度上起到了肺保護的作用。
本部分研究主要發現:體外培養的PMVEC 在給予腦缺血-再灌注大鼠滅活的血清和外源性ADMA干預后,由于PMVEC 培養液中ADMA 含量增高,PKCα、MLCK 蛋白和mRNA 表達增加,細胞增殖和生長明顯下降。這些發現提示ADMA/DDAH 通路在腦缺血-再灌注損傷所致ALI 可能起著重要作用,可以作為防治缺血再灌注藥物新的潛在的干預靶點,研究藥物干預ADMA 的生成代謝,干預ADMA/DDAH 系統,上調DDAH 的表達,抑制ADMA 的生成,因此推測藥物干預ADMA 的代謝或將是防治缺血再灌注疾病導致ALI 的一個新的治療目標。
[1]Antohe F.Endothelial cells and macrophages,partners in atherosclerotic plaque progression[J].Arch Physiol Biochem,2006,112(4):245-253.
[2]Kharitonova MA,Levina CM,Rovenskii IA.Cytoskeletal control of cell length regulation[J].Ontoqenez,2002,33(1):50-59.
[3]Benz PM,Blume C,Moebius J,et al.Cytoskeleton assembly at endothelial cell-cell contacts is regulated by alphaII-spectrin-VASP complexes[J].Cell Biol,2008,180:205-219.
[4]Mellor H,Parker P.The extended protein kinase C superfamily[J].Biochem,1998,332:281-292.
[5]Ron D,Kazanietz MG.New insight into the regulation of protein kinase C and novel phorbol cster receptors[J].FASEB,1999,13:1658-1678.
[6]Furman,C,Sieminski AL,Kwiatkowski A,et al.Ena/VASP is required for endothelial barrier function in vivo[J].Cell Biol,2007,179:761-775.
[7]Stull JT,Kamm KE,Krueger JK,et al.Ca2+/calmodulin-dependent myosin light chain kinases[J].Adv Second Messenger Phosphoprotein Res,1997,31:141-150.
[8]Klinger JR,Warburton R,Carino S,et al.Natriuretic peptides differentially attenuate thrombin-induced barrier dysfunction in pulmonary microvascular endothelial cells[J].Exp Cell Res,2006,15:401-410.
[9]陳 鐸,石玉秀,西澤茂,等.實驗性腦血管痙攣中PKC 活性表達及其意義[J].中國醫科大學學報,2000,29:91-93.
[10]Boushey CJ,Beresford SAA,Omen GS,et al.A quantitative assessment of plasma homocysteine as a risk factor for vascular disease[J].JAMA,1995,274:1049-1053.
[11]Schlegel N,Burger S,Golenhofen N,et al.The role of VASP in regulation of cAMP-and Rac 1-mediated endothelial barrier stabilization[J].Am Physiol Cell Physiol,2008,294:178-188.
[12]Wojciak-Stothard B,Torondel B,Tsang LY,et al.The ADMA/DDAH pathway is a critical regulator of endothelial cell motility[J].Cell Sci,2007,120:929-942.
[13]Kwiatkowski A.Pressure-related increase of asymmetric dimethylarginine caused by hyperbaric oxygen in the rat brain:a possible neuroprotective mechanism[J].Cell Biol,2009,171:762-778.
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
中老年保健(2021年3期)2021-08-22 06:50:04
昆明醫科大學學報(2021年1期)2021-02-07 01:06:36
現代臨床醫學(2021年1期)2021-01-26 00:56:02
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
中華養生保健(2020年4期)2020-11-16 01:31:40
中西醫結合肝病雜志(2020年2期)2020-10-27 02:18:50
數學物理學報(2020年2期)2020-06-02 11:29:24
光學精密工程(2016年6期)2016-11-07 09:07:19
核科學與工程(2015年4期)2015-09-26 11:59:03
