視紫紅質和Peripherin/RDS基因在視網膜色素變性家系中的突變檢測
2012-10-11 08:19:18章雪敏劉鐵城張寶全
解放軍醫學院學報 2012年6期
章雪敏,劉鐵城,金 鑫,張寶全,徐 華
1解放軍總醫院 眼科,北京 100853;2四川省宣漢縣人民醫院 眼科,達州 636150
原發性視網膜色素變性(retinitis pigmentosa,RP)是視網膜感光細胞和色素上皮細胞變性導致夜盲和進行性視野損害的遺傳性致盲眼病。我國發病率約為1/3 784[1]。RP的遺傳方式復雜多樣,非綜合征RP至少有4種遺傳方式,其中15%-25%為常染色體顯性遺傳(adRP),5%-20%為常染色體隱性遺傳 (autosomal recessive RP,arRP),5%-15%為X-連鎖遺傳(X-linked RP,XLRP),另有5%早發型RP即隱性Leber's先天性黑蒙(LCAⅡ型)。當患者因缺乏家族史無法確定其遺傳方式時,將其定義為散發型RP(sporadic RP,SRP),約占40%-50%[2]。多數RP為單基因遺傳,但也存在二基因-雙等位基因和二基因-三等位基因遺傳方式[3]。迄今為止,發現與RP有關的致病基因共59個,其中adRP 23個[4]。本研究對一個常染色體顯性視網膜色素變性家系成員進行RHO、Peripherin/RDS、ROM1、NRL和CRX 5個常見候選基因檢測,在分子水平闡述該家系的發病機制。
對象和方法
1 對象 本研究家系來自四川省達州市普光縣,5代46名成員,已采集外周血的成員為28名,其中6名RP患者、1名疑似成員和21名正常成員。該家系RP性狀連續3代傳遞,男女均可發病,發病比例為1∶5,符合常染色體完全顯性遺傳特征。
2 臨床資料采集 按照RP診斷標準[5]詢問家系成員病史,繪制家系圖譜(圖1),確定疾病遺傳類型,所有研究對象均行視力、眼壓、房角、晶狀體、眼底、視野及視網膜電圖(electroretinography,ERG)等檢查。分析家系的臨床特點:患者年齡33-79歲,出現夜盲時間為5-15歲。視野損害出現時間各異,患者II:1、Ⅲ:1和Ⅳ:1無視野縮小病史。Ⅲ:1和Ⅲ:3出現紅色覺減弱。Ⅲ:5和Ⅲ:7虹膜膨隆,房角為窄角Ⅲ-Ⅳ級,但眼壓均正常。Ⅲ:1、Ⅲ:3、Ⅲ:5和Ⅲ:7出現并發性白內障。Ⅲ:1和Ⅲ:3玻璃體呈煙灰樣混濁。除II:1外,所有患者黃斑區均出現骨細胞樣色素沉著。疑似成員Ⅴ:1為11歲男性,父母訴其5歲出現夜盲,眼部檢查未見異常。采集到血樣的21名正常成員為Ⅲ:2、Ⅲ:4、Ⅲ:6、Ⅲ:9、Ⅳ:2-Ⅳ:6、Ⅳ:12、Ⅳ:13、Ⅳ:17、Ⅳ:18、Ⅴ:2-Ⅴ:9,眼底均未見異常。
3 外周血DNA制備 本研究通過解放軍總醫院倫理委員會論證,所有受檢者簽署知情同意書。抽取該家系中6名患者、1名疑似成員和21名正常成員外周靜脈血8-10ml,EDTA抗凝。DNA抽提采用天根生化科技(北京)有限公司提供的離心柱型血液基因組DNA抽提試劑盒,按操作方法提取外周血白細胞DNA,-80℃保存待用。
4 PCR擴增 采用Primer3軟件在線設計RHO、Peripherin/RDS、ROM1、NRL和CRX基因的 16個外顯子及其與內含子交界處序列的引物[6-8],引物由北京奧科生物技術公司合成。PCR反應在25μl體系中進行:2×Taq PCR MasterMix(含染料,藍色)12.5μl,正反向引物各5μl(引物原液加滅菌ddH2O稀釋50倍),DNA模板2.5μl(模板原液)。反應條件:94℃ 3min,94℃ 30s,55℃ 30s,72℃1min,共30個循環,72℃延伸5min。PCR產物經1%瓊脂糖凝膠電泳,溴化乙錠/紫外線鑒定,證實PCR擴增片段存在。
5 測序結果分析 將所得PCR產物送至北京中美泰和生物技術有限公司測序,所得結果使用Chromas軟件、GeneTool軟件和在線BLAST程序(http//www.ncbi.nlm.govBLAST)進行閱讀和堿基序列比對。
結 果
該家系患者擴增產物在RHO、Peripherin/RDS、ROM1、NRL和CRX基因中未發現致病突變,但在Peripherin/RDS基因第1外顯子(exon)和第3外顯子編碼區發現4處單核苷酸改變,屬于單核苷酸多態性(single nucleotide polymorphisms,SNPs)。在正常成員中,這些部位的氨基酸均和美國國立生物技術信息中心(National Center for Biotechnology Information,NCBI)數據庫上公布的標準序列一致。如表1所示。相對應的峰圖見圖2。
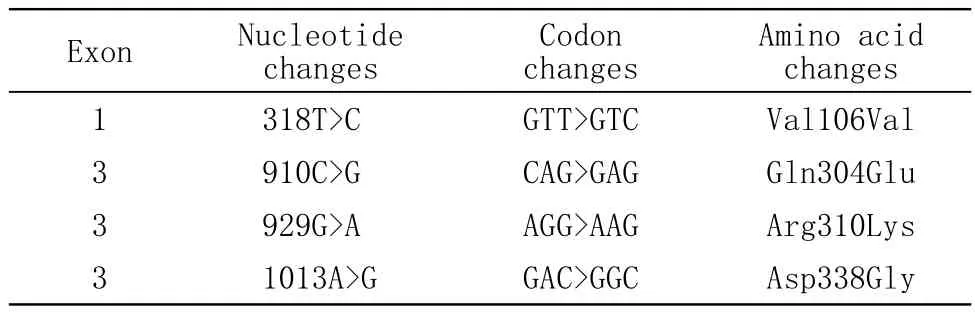
表1 Peripherin/RDS基因測序結果Tab 1 Peripherin/RDS gene sequencing
討 論
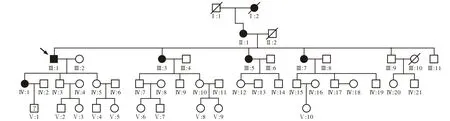
圖1 adRP家系圖譜系譜圖注釋:Fig 1 Pedigree of adRPNote:

圖2 SNPs測序峰圖Fig 2 Sequencing picture of SNP
據報道[9],RHO和Peripherin/RDS基因分別占adRP致病基因的25%-30%和5%-10%,因此,本研究首先選取ROH(Exon1-5)、Peripherin/RDS(Exon 1-3)、ROMI(Exon1-3)、CRX(Exon2-4)和NRL(Exon2、3)基因的16個外顯子進行初步篩查,結果發現在Peripherin/RDS基因上存在4處SNPs:910C>G、929G>A和1013A>G為 錯 義 突 變, 對應的氨基酸改變為:Gln304Glu、Arg310Lys和Asp338Gly;318T>C為同義突變。其余候選基因未發現堿基改變。
RHO基因定位于3q21-q24,由5個外顯子和4個內含子組成,長約6952bp,編碼由348個氨基酸組成的視紫紅質(rhodopsin,RHO)蛋白,該蛋白僅在視桿光感受器細胞中專一性表達,與RP發病早期即存在視桿細胞變性相一致[10]。目前,已發現100余種[6]RHO突變與RP有關,在這些突變譜中,90%以上是單個堿基突變,少數是小片段的插入或缺失突變(Indel),一般不超過20個堿基。研究表明,Pro23His和Pro347Leu突變在歐美最為常見,Pro347Leu突變在我國較常見[11]。RHO基因突變率存在地區差異,我國低于歐美,約占adRP致病基因的7%[12]。本研究RHO基因未發現堿基改變,確定該基因與本研究adRP家系臨床表型無關。
Peripherin/RDS 基因位于 6p21[13],cDNA長 3 212個堿基,由3個外顯子和2個內含子組成,編碼由346個氨基酸組成的盤膜周邊蛋白,表達于光感受器細胞內,其功能可能與光感受器的發生有關,并且維持外節盤緣的結構及其穩定性。Peripherin/RDS基因突變最重要的特點是疾病表型多樣化,目前,公認的觀點是由于修飾位點的存在和環境因素的影響所致[14]。Peripherin/RDS基因突變引起的表型改變可以分為四類:ADRP、進行性黃斑變性、雙基因RP和視網膜圖形樣變性,該基因內不同位點和突變類型引起疾病的嚴重程度不同:第三和四跨膜區之間的錯義突變和讀框缺失引起ADRP和嚴重黃斑變性;分布于整個Peripherin/RDS基因的無意義突變引起圖形樣黃斑變性和雙基因RP。本研究未發現該家系在Peripherin/RDS基因中存在致病突變,但在Peripherin/RDS基因外顯子1和3編碼區發現4處SNPs,均不與疾病共分離。說明Peripherin/RDS基因存在遺傳異質性。
本研究家系患者臨床表型差異性明顯,其原因可能是由于核苷酸堿基的單倍體不同,或順式或反式等位基因不同,也可能是環境因素影響了致病基因的突變,從而影響疾病的臨床表型[15]。現通過直接測序的方法已排除RHO、Peripherin/RDS、ROM1、NRL和CRX基因與該家系臨床表型相關,Peripherin/RDS基因外顯子區發現的4處SNPs,與dbSNP數據庫中(http://www.ncbi.nlm.nih.gov/SNP/)的記錄進行比對,未發現與本研究相同的SNP位點,因此,本研究發現的SNPs有可能是新的SNP位點。我們下一步的工作是,繼續對剩余的18個adRP候選基因進行檢測,鑒于用Sanger測序法工作量大,所需時間長,因此采用高通量測序技術進行后續工作(另文報告)。本家系表型差異性大,不排除存在新的致病基因的可能。
1 覃泳杰,郭海科. 常染色體遺傳型視網膜色素變性相關基因的研究進展[J]. 眼科研究,2009,27(12):1159-1163.
2 Daiger SP,Bowne SJ,Sullivan LS. Perspective on genes and mutations causing retinitis pigmentosa[J]. Arch Ophthalmol,2007,125(2):151-158.
3 Kajiwara K,Berson EL,Dryja TP. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci[J].Science,1994,264(5165):1604-1608.
4 Summaries of Genes and Loci Causing Retinal Diseases[EB/OL].https://sph.uth.tmc.edu/RetNet/sum-dis.htm.
5 莊文娟,鄧大君,盛迅倫,等. 常染色體顯性遺傳RP患者視紫紅質基因突變 的檢測分析[J]. 眼科研究,2006,24(4):348-351.
6 劉逾,賈曉林,李進軍,等. 視網膜色素變性患者視紫紅質基因突變分析[J]. 中國全科醫學,2011,14(15):1659-1662.
7 Matias-florentino M,Ayala-ramirez R,Graue-wiechers F,et al.Molecular screening of rhodopsin and peripherin/RDS genes in mexican families with autosomal dominant retinitis pigmentosa[J].Curr Eye Res,2009,34(12):1050-1056.
8 Ferrari S,Di IE,Barbaro V,et al. Retinitis pigmentosa:genes and disease mechanisms[J]. Curr Genomics,2011,12(4):238-249.
9 Pagon RA,Daiger SP. Retinitis pigmentosa overview[M].Washington:Gene Clinics,2005.
10 Zhang XL,Liu M,Meng XH,et al. A complete screen for mutations of the rhodopsin gene in a panel of Chinese patients with autosomal dominant retinitis pigmentosa[J]. Chin Med Sci J,2005,20(1):30-34.
11 Zhang XL,Liu M,Meng XH,et al. Mutational analysis of the rhodopsin gene in chinese ADRP families by conformation sensitive gel electrophoresis[J]. Life Sci,2006,78(13):1494-1498.
12 管濤,麻張偉,丁世萍. 散發性視網膜色素變性視紫紅質基因突變篩查[J]. 中國眼耳鼻喉科雜志,2008,8(6):351-353.
13 Travis GH,Christerson L,Danielson PE,et al. The human retinal degeneration slow(RDS)gene:chromosome assignment and structure of the mRNA[J]. Genomics,1991,10(3):733-739.
14 周占宇,嚴密,陳大年. 盤膜邊緣蛋白和桿體外節盤膜蛋白與視網膜光感受器 變性[J]. 中華眼底病雜志,1999,15(3):197.
15 盛迅倫. 視網膜色素變性的遺傳異質性和臨床異質性[J]. 山東大學耳鼻喉眼學報,2011,25(5):94-98.
