手性氨基亞磺酰胺的合成及其對N-苯基酮亞胺不對稱還原的催化作用*
2011-11-26 01:49:44盧曉霞
合成化學 2011年1期
王 萌, 王 超, 盧曉霞, 孫 健
(1. 中國科學院 成都生物研究所 天然產物研究中心,四川 成都 610041;2. 中國科學院 研究生院,北京 100049)
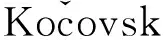
Scheme 1
本文在前期工作[10~16]的基礎上,從手性α-氨基醇(1a,1b和1f)和(R)-叔丁基亞磺酰胺(Ⅰ)出發,進一步設計并合成了一系列新型的同時含有C-手性和S-手性的氨基亞磺酰胺有機小分子催化劑(5a~5f, Scheme 1),其結構經1H NMR和MS表征。將5應用于N-苯基酮亞胺的不對稱硅氫化反應中作催化劑,獲得了較好的結果。
1 實驗部分
1.1 儀器與試劑
Brucker-600型和300型核磁共振儀(CDCl3為溶劑,TMS為內標);Waters Q-tof Premier型質譜儀;Agilent Technologies 1200 Series型高效液相色譜儀(手性柱AS-H, AD-H, OD-H 和OJ-H)。
所用試劑均為分析純;柱層析使用硅膠H,青島海洋化工廠。
1.2 合成
(1)N-Fmoc-L-脯胺醛(2a)的合成
在反應瓶中加入N-Fmoc-L-脯胺醇(1a) 2 g(6.2 mmol)的乙酸乙酯(50 mL)溶液和鄰-碘酰基苯甲酸5.2 g(18.5 mmol),攪拌下于85 ℃反應3 h。冷卻至室溫,過濾,濾液濃縮得粗品2a(2 g)直接用于下步反應。
(2) (R)-N-Fmoc-L-脯胺叔丁基亞磺酰基亞胺(3a)的合成
在反應瓶中加入2a1.1 g(3.43 mmol), Ⅰ 500 mg(4.11 mmol)和二氯甲烷20 mL,攪拌下加入無水CuSO41.1 g(6.9 mmol), 于室溫反應48 h。過濾,濾液減壓濃縮后經硅膠柱層析[洗脫劑: A=V(石油醚) ∶V(乙酸乙酯)=3 ∶1]純化得淡黃色固體3a1.4 g,產率96%。
(3) (R)-N-Fmoc-L-脯胺叔丁基亞磺酰胺(4a)的合成
在反應瓶中加入3a1.4 g(3.3 mmol)和甲醇15 mL,攪拌下于至0 ℃加入 NaBH4250 mg(6.6 mmol),于室溫反應5 h。加入丙酮(1 mL)淬滅反應,蒸除溶劑,殘余物用飽和氯化銨溶液洗滌,二氯甲烷(3×20 mL)萃取,合并有機相,用飽和食鹽水洗滌,無水MgSO4干燥,減壓濃縮得粗品4a(1.4 g),直接用于下步反應。
(4) (R)-L-脯胺叔丁基亞磺酰胺(5a)和(R)-α-哌嗪叔丁基亞磺酰胺(5b)的合成
在反應瓶中加入4a1.4 g(3.3 mmol)和THF 20 mL,攪拌下加入二乙基胺4 mL,于室溫反應6 h。減壓濃縮后經硅膠柱層析[洗脫劑:B=V(二氯甲烷) ∶V(甲醇)=15 ∶1]純化得黃色固體5a350 mg,產率52%;1H NMRδ: 1.19(s, 9H), 1.37~1.41(m, 1H), 1.69~1.83(m, 3H), 2.30(s, 1H), 2.88~2.92(m, 3H), 3.18~3.23(m, 1H), 3.38~3.41(m, 1H), 3.90(brs, 1H); ESI-HR-MSm/z: Calcd for [C9H20N2OS+H]+205.129 6, found 205.138 2。
以1b代替1a,用類似方法[1.2(1)~1.2(4)]合成黃色固體5b[洗脫劑:B=40 ∶1] 350 mg,產率52%;1H NMRδ: 1.17~1.18(m, 1H), 1.21(s, 9H), 1.35~1.39(m, 2H), 1.58~1.59(m, 2H), 1.62~1.63(m, 1H), 2.24(br s, 1H), 2.62~2.69(m, 2H), 2.94~2.98(m, 1H), 3.06~3.08(m, 1H), 3.19~3.23(m, 1H),3.59(t,J=5.62 Hz, 1H); ESI-HR-MSm/z: Calcd for [C10H22N2OS+H]+219.145 3, found 219.153 7。
(5) (R)-N-甲酰-L-脯胺叔丁基亞磺酰胺(5c)和(R)-N-甲酰-α-哌嗪叔丁基亞磺酰胺(5d)的合成
在反應瓶中加入5a150 mg(0.75 mmol)和甲酸甲酯(5 mL),攪拌下于室溫反應12 h。減壓濃縮后經硅膠柱層析(洗脫劑:B=40 ∶1)純化得淺黃色固體5c127 mg,產率75%;1H NMRδ: 1.18(s, 9H), 1.72~1.91(m, 4H), 2.35(br s, 1H), 3.17~4.14(m, 4H), 4.68(t,J=5.62 Hz, 1H), 8.23(s, 1H); ESI-HR-MSm/z: Calcd for [C10H20N2O2S+H]+233.124 5, found 233.132 7。
以5b代替5a,用類似方法合成淺黃色固體5d(洗脫劑:B=40 ∶1) 127 mg,產率75%;1H NMRδ: 1.13(s, 9H), 1.33~1.69(m, 6H), 1.83(br s, 1H), 3.08~3.19(m, 2H), 3.31~3.75(m, 1H), 3.55~3.57(m, 1H), 4.53~4.55(m, 1H), 8.01(s, 1H); ESI-HR-MSm/z: Calcd for [C11H22N2O2S+H]+247.140 2, found 247.147 9。
(6) (R)-N-芐基-L-脯胺叔丁基亞磺酰胺(5e)的合成
在反應瓶中加入5a150 mg(0.75 mmol), 1,2-二氯乙烷6 mL和苯甲醛87 mg(0.83 mmol),攪拌下加入NaBH(OAc)3200 mg(0.9 mmol),于室溫反應24 h。加入丙酮 (1 mL)淬滅反應。蒸除溶劑,用飽和NH4Cl水溶液洗滌,乙酸乙酯(3×20 mL)萃取,合并有機相,用飽和食鹽水洗滌,無水MgSO4干燥,減壓濃縮后經硅膠柱層析[洗脫劑:B=40 ∶1]純化得淺黃色固體5e220 mg,產率83%;1H NMRδ: 1.23(s, 9H), 1.64~1.71(m, 2H), 1.88~1.92(m, 1H), 2.20~2.26(m, 2H), 2.79(br s, 1H), 2.96~3.04(m, 2H), 3.30~3.42(m, 2H), 4.01(s, 1H), 4.06(s, 1H), 7.22~7.31(m, 5H); ESI-HR-MSm/z: Calcd for [C16H26N2OS+H]+295.176 6, found 295.184 8。
(7) (R)-N-甲基-L-脯胺叔丁基亞磺酰胺(5f)的合成
在反應瓶中加入草酰氯1.4 mL(16.1 mmol)和二氯甲烷80 mL,攪拌下于-78 ℃加入DMSO 2.05 mL(28.98 mmol)的二氯甲烷(24 mL)溶液,反應15 min;緩慢滴加N-Cbz-L-脯胺醇(1f) 1.5 g(6.44 mmol)的二氯甲烷(21 mL)溶液,滴畢,反應30 min;滴加三乙胺4.78 mL(32.2 mmol),滴畢,用水(10 mL)淬滅反應,減壓濃縮后用乙酸乙酯(3×45 mL)萃取,合并有機層,依次用水(15 mL),飽和食鹽水(5 mL)洗滌,無水Na2SO4干燥,減壓濃縮后經硅膠柱層析(洗脫劑: A=10 ∶1)純化得無色油狀液體N-Cbz-L-脯胺醛(2f) 1.33 g,產率90%。
以2f代替2a,用類似方法[1.2(2), 1.2(3)]合成淺黃色油狀液體(R)-N-Cbz-L-脯胺叔丁基亞磺酰基亞胺(3f, 洗脫劑:A=10 ∶1)和淺黃色油狀液體(R)-N-Cbz-L-脯胺叔丁基亞磺酰胺(4f, 洗脫劑: A=1 ∶1)。
在反應瓶中加入4f372 mg(1.14 mmol)和THF 6 mL,攪拌下于0 ℃加入LiAlH4100 mg(2.63 mmol),于70 ℃反應3 h。用水(0.1 mL)淬滅反應,依次加入15%NaOH溶液(0.1 mL)和水(0.3 mL),過濾,濾液減壓濃縮后經硅膠柱層析(洗脫劑:B=20 ∶1)純化得淺黃色固體5f62 mg,產率25%;1H NMRδ: 1.21(s, 9H), 1.67~1.74(m, 2H), 1.85~1.87(m, 1H), 2.23~2.28(m, 2H), 2.33(s, 3H), 2.45(br s, 1H), 3.02~3.06(m, 2H), 3.27~3.32(m, 1H), 3.90(br s, 1H); ESI-HR-MSm/z: Calcd for [C10H22N2OS+H]+219.145 3, found 219.153 2。
1.3 5催化的N-苯基酮亞胺的不對稱硅氫化反應
在卷口試管(10 mL)中加入5 0.02 mmol,N-苯基酮亞胺0.1 mmol和二氯甲烷1 mL,攪拌下于-20 ℃加入HSiCl30.04 mL,反應24 h。依次用飽和氯化銨(1 mL)溶液,飽和碳酸氫鈉溶液(1 mL)淬滅反應,用乙酸乙酯(3×15 mL)萃取,合并有機相,用無水MgSO4干燥,濃縮后經柱層析(洗脫劑:A=20 ∶1)純化得淺黃色油狀液體N-苯基手性胺。
2 結果與討論
2.1 5的合成
從N-保護的手性氨基醇出發,通過氧化成醛后,與(R)-叔丁基亞磺酰胺發生縮合反應得到叔丁基亞磺酰基亞胺化合物,再經過一步還原胺化合成了一系列新型的同時含有C-手性和S-手性的氨基亞磺酰胺有機小分子催化劑。
2.2 5催化的N-苯基酮亞胺的不對稱硅氫化反應
以N-苯基酮亞胺的不對稱硅氫化反應(Scheme 2)為模板,反應條件同1.3,考察5的催化性能,實驗結果見表1。從表1可以看出,5均能催化N-苯基酮亞胺的不對稱硅氫化反應。其中,以五元環為骨架的5a和5c的催化效果優于以六元環為骨架的5b和5d。另外,5a和5b環上的氮原子被甲酰化后(5c和5d),其催化活性有較大提高(收率>90%),但是對映選擇性卻明顯降低(<50%)。此外,我們發現當5a的氮原子上帶有芳香基團如芐基時(5e),其立體控制作用增強,但催化效果較差;然而若將芐基替換成甲基時(5f),其對映選擇性與5a相當,并且催化效果有所提高。總體來講,5f表現最為優秀,其催化還原產物N-苯基手性胺可以達到68%的收率和69%的對映選擇性。
Scheme 2
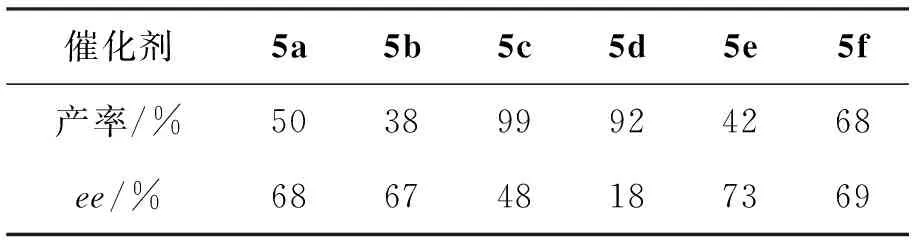
表1 5催化的N-苯基酮亞胺的不對稱硅氫化反應
3 結論
本文首次從手性氨基醇和手性叔丁基亞磺酰胺出發設計并合成一類新型路易斯堿催化劑,并將其應用于酮亞胺的不對稱硅氫化反應,獲得了中等的收率與對映選擇性。
[1] Vilaivan T, Bhanthumnavin W, Sritana-Anant Y. Recent advances in catalytic asymmetric addition to imines and related C=N systems[J].Curr Org Chem,2005,9:1315-1392.
[2] Blaser H U, Malan C, Pugin B. Selective hydrogenation for fine chemicals:Recent trends and new developments[J].Adv Synth Catal,2003,345:103-151.
[3] Tang W, Zhang X. New chiral phosphorus ligands for enantioselective hydrogenation[J].Chem Rev,2003,103:3029-3069.
[4] Adams J P. Imines,enamines and oximes[J].J Chem Soc Perkin Trans,2000,1:125-139.
[5] Kobayashi S, Ishitani H. Catalytic enantioselective addition to imines[J].Chem Rev,1999,99:1069-1094.
[6] Bloch R. Additions of organometallic reagents to C=N bonds: reactivity and selectivity[J].Chem Rev,1998,98:1407-1438.
[7] Malkov A V, Mariani A, MacDougall K N. Role of noncovalent interactions in the enantioselective reduction of aromatic Ketimines with trichlorosilane[J].Org Lett,2004,6:2253-2256.
[8] Malkov A V, Stewart Liddon A J P, Ramírez-López P. Remote chiral induction in the organocatalytic hydrosilylation of aromatic ketones and ketimines[J].Angew Chem Int Ed,2006,45:1432-1435.
[9] Malkov A V, Stoncius S, Kocovsky P. Enantioselective synthesis of 1,2-diarylaziridines via organocatalytic reductive amination ofα-chloro ketones[J].Angew Chem Int Ed,2007,46:3722-3725.
[10] Wang Z, Sun J. A highly enantioselective lewis basic organocatalyst for reduction ofN-aryl imines with unprecedented substrate spectrum[J].Org Lett,2006,8:999-1001.
[11] Wang Z, Sun J. L-piperazine-2-carboxylic acid derivedN-formamide as highly enantioselective Lewis basic catalyst for hydrosilylation ofN-aryl imines with an unprecedented substrate profile[J].Org Lett,2006,8:3045-3048.
[12] Wang Z, Sun J. Enantioselective hydrosilylation of ketimines catalyzed by Lewis basicC2-symmetric chiral tetraamide[J].Tetrahedron: Asymmetry,2007,18:705-709.
[13] Zhou L, Sun J. Evolution of chiral Lewis basicN-formamide as highly effective organocatalyst for asymmetric reduction of both ketones and ketimines with an unprecedented substrate scope[J].Chem Commun,2007:2977-2979.
[14] Pei D, Sun J.S-chiral sulfinamides as highly enantioselective organocatalysts[J].Org Lett,2006,8:5913-5915.
[15] Pei D, Sun J. Rationally-designedS-chiral bissulfinamides as highly enantioselective organocatalysts for reduction of ketimines[J].Adv Synth Catal,2008,350:619-623.
[16] Wang C, Sun J. A highly enantioselective organocatalytic method for reduction of aromaticN-alkyl ketimines[J].Chem Eur J,2008,14:8789-8792.
