微波輔助合成咪唑類化合物的研究
2011-05-18 09:10:54孟江平徐強宋仲容陳鳳鳴何家洪
重慶高教研究 2011年3期
關鍵詞:方法
孟江平,徐強,宋仲容,陳鳳鳴,何家洪
(1.重慶文理學院化學與環境工程學院,重慶 永川 402168;2.重慶市高校微納米材料工程與技術重點實驗室,重慶 永川 402160)
微波在化學中的應用開辟了微波化學這個化學新領域[1].微波可以與化學體系發生作用從而促進各類化學反應的進行.它對化學反應作用是非常復雜的,一方面是反應物分子吸收了微波能量,提高了分子運動速度,致使分子運動雜亂無章,導致熵的增加;另一方面,微波對極性分子的作用,迫使其按照電磁場作用方式運動,每秒變化2.45×109次,導致了熵的減小.此外,它還存在一種不是由溫度引起的非熱效應.微波作用下的有機反應,改變了反應動力學,降低了反應活化能.近年來,大量的實驗已經證實,微波可以極大地提高一些化學反應的反應速率,使一些在通常條件下不易進行的反應迅速形成.傳統上,有機合成或藥物合成采用外部熱源進行傳導加熱,如油浴或加熱套等.但是傳統的加熱方法多是間接加熱,并受制于對流和必須透過的各種材料的導熱性,相對而言它在向體系傳輸能量時速度緩慢,效率不高,而且導致反應容器的溫度高于反應混合物的溫度.此外,被加熱底物內部會產生溫度梯度,而且反應物局部過熱也可導致產物、底物或者試劑的分解.相反,通過微波能量和反應混合物中的分子如溶劑、試劑或催化劑的直接耦合,可以直接進行充分的內部加熱[2].
咪唑環是生物體內組胺、組氨酸產生生物活性和發揮生理作用的重要基團[3-5].咪唑環是結構中含有2個氮原子的五元芳氮雜環,易產生多種非共價鍵相互作用,如氫鍵、與金屬離子配位和π-π作用等[6-8].以這種特殊結構的咪唑環構筑的咪唑類衍生物具有較大的發展潛力,如作為人工受體用于分子識別[9],作為人工酶用于仿生催化[10],作為藥物具有廣泛的生物活性,如組胺受體拮抗劑[11]、酶抑制劑[12]、質子泵抑制劑[13]、抗微生物[14]、抗癌[15]等等.因此,近年來咪唑類化合物的合成與開發越來越受到人們的重視.本文結合自己的研究工作,就近年來有關微波輻射合成咪唑類化合物的國內外相關情況進行綜述.
1 微波輔助1,2-二酮和醛合成咪唑類化合物
通過1,2-二酮與醛及其衍生物的反應合成咪唑及其類似物是一個相當傳統的合成路徑.傳統的方法往往需要較為苛刻的條件——強酸性條件,較高的溫度,很長的時間等.近年來,隨著微波技術在有機合成中的廣泛應用,極大地改善了用1,2-二酮與醛及其衍生物合成咪唑的條件.微波輻射1,2-二酮與醛合成咪唑具有速度快、產率高、污染少、安全性高等優點.
Wolkenbergd等[16]在乙酸鋁的存在下,由 1,2-二酮和醛簡單而高產率的合成了2,4,5-三取代咪唑3.微波照射,溫度為180℃,在乙酸鋁的存在下,5min之內,1,2-二酮和醛縮合合成烷基、芳基和雜芳基取代的咪唑衍生物,產率可達76%~99%.在三乙胺堿性條件下,2,4,5-三甲基咪唑與芐氯進一步進行微波輔助烷基化反應,以43%的總產率得到了生物堿勒皮啶(lepidine)4.帶有不對稱咪唑鎓結構的勒皮啶4在微摩爾級顯示了抗多種人腫瘤細胞株的細胞毒性,該化合物作為臨床抗腫瘤藥物具有進一步開發價值.值得注意的是,中間體2,4,5-三甲基咪唑的制備在技術上簡便、快速,與以前的路徑相比產率更高.化合物3和4的合成路線如圖1所示.
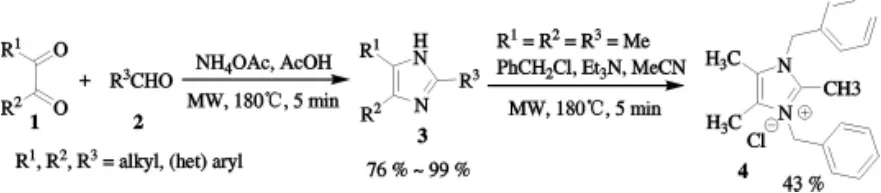
圖1 化合物3和4的合成路線
2 微波輔助酮肟和醛合成咪唑類化合物
Sparks和Combs等[17]研究了以非對稱酮肟與不同的醛為原料,利用微波輔助合成咪唑類化合物的方法,按照這一方式高產率地合成了N-羥基咪唑7.隨后以三氯化鈦為催化劑,將其定量還原為咪唑化合物8,反應時間僅需5min.研究發現,在高反應溫度如200℃下處理酮肟與醛及乙酸銨時,經原位切斷熱易變的N—O鍵直接得到了目標化合物8.利用這種優化的條件可制備多樣化、高效率的2,4,5-三(雜)芳基咪唑,給咪唑環的合成帶來了新的方法.化合物7和8的合成路線如圖2所示.
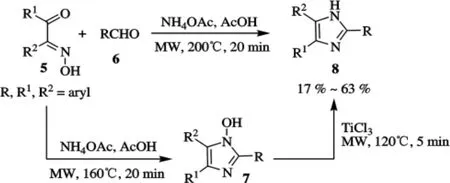
圖2 化合物7和8的合成路線
3 微波輔助多組分反應(MCRs)合成咪唑類化合物
多組分反應(Multicomponent Reactions,MCRs)是指3個或3個以上的起始原料進入反應,用“一鍋煮”的方法最終生成一個終產物,在終產物結構中含有所有原料片段的合成方法[18].多組分反應由于其高度的原子經濟性、收斂性、出色的產率等特性以及在藥物研究中先導物的發現和結構優化等方面的重要應用,在有機和藥物化學領域發揮著越來越重要的作用.應用多組分反應構筑新的、具有各種生理活性的雜環化合物已成為雜環合成的重要手段之一[19-20].
Ugi四組分縮合反應,即胺、醛和酮、羧酸以及異腈結合產生α-酰基氨的反應是目前有機合成中的常用方法,因其通過不同的起始原料可以獲得范圍很寬的產物而具有特別意義.在以三氟甲磺酸鈧為催化劑進行的Ugi型三組分縮合反應中,雜環脒與醛和異腈在室溫下反應通常需要長達72 h的反應時間才能生成期望的稠合3-氨基咪唑.Ireland等[21]在微波條件下合成了咪唑類化合物12,合成路線如圖 3所示.在160℃下以甲醇作為溶劑,在有些例子中采用乙醇為溶劑,10min就可以產生與室溫相同過程類似的產率,而時間只是室溫反應的一小部分,極大地提高了合成效率,縮短了反應時間.
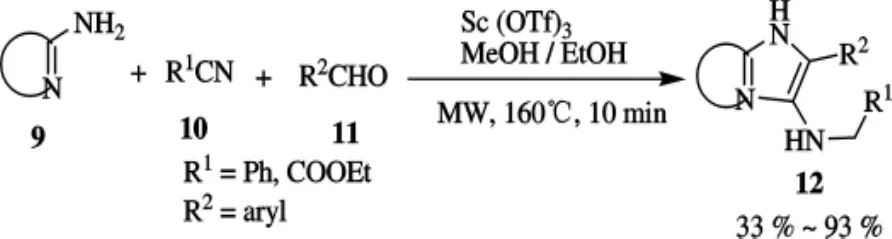
圖3 化合物12的合成路線
在乙酸銨存在下,醛、2-氧代硫代乙酰胺以及溴代烷之間的具有多種包容性三組分縮合合成咪唑類化合物[22].利用微波輔助多組分反應原理,以結構不同的醛,溴代烷和2-氧代硫代乙酰胺為原料,合成了咪唑類化合物16,合成路線如圖4所示.生物活性研究表明,16是一類潛在的酰基輔酶A/膽固醇酰基轉移酶抑制劑、止痛劑以及血管緊張素受體的拮抗劑.
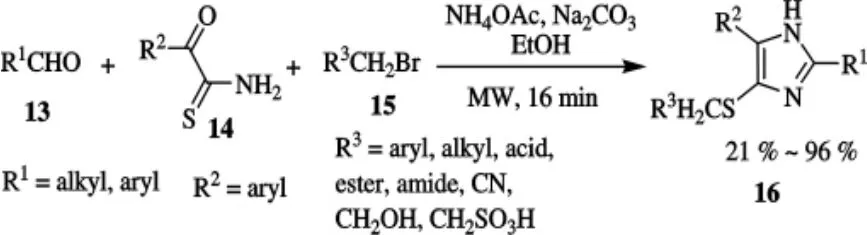
圖4 化合物16的合成路線
4 微波輔助合成咪唑啉及其衍生物
咪唑啉是咪唑的重要衍生物之一,在醫藥、催化劑、洗滌品及緩蝕劑等領域具有廣泛用途.傳統的合成方法是采用不同種類的羧酸、酯、腈或酰氯等與乙二胺或多胺類化合物反應制得.這一傳統的方法具有耗時長、產率低、分離困難及產物成分復雜等缺點.微波輔助技術極大地改善了這些缺點,為咪唑啉類化合物的高通量合成帶來了光明的前景.
劉金艷等[23]以吡啶2,6-二甲酸與乙二胺為原料,合成了吡啶咪唑啉類化合物20,合成路線如圖5所示.這一合成方法顯著縮短了反應時間,提高了產率,減少了環境污染.反應過程中,微波功率為450 W,輻射時間為65min時產率最好.
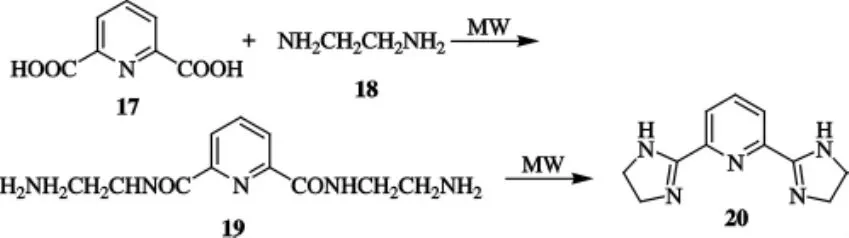
圖5 化合物20的合成路線
Merriman等[24]報道了由固相方法得到N-酰基-1,2-二芳基-1,2-乙烷二胺的環化,通過在二氯甲烷溶液中以三甲硅基多聚磷酸鹽(TMS-PP)處理后,生成了4,5-二芳基咪唑啉22,合成路線如圖6所示.由微波在140℃下加熱8 min獲得了最佳結果.以這種方法制備了含有38個化合物的庫,產生了一個新且有效的P2X7受體拮抗劑家族.
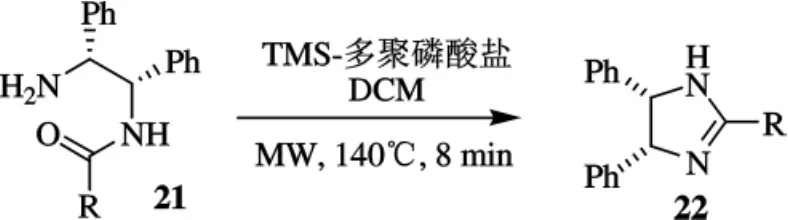
圖6 化合物22的合成路線
在氧化鋅的存在下,以DMF為溶劑,在微波照射120℃下亞乙基二胺和脲進行縮合,以95%的分離產率得到了咪唑啉-2-酮25,產率高達95%,合成路線如圖7所示.這一方法成功的關鍵之處在于,反應需要在負壓下完成,以便從反應混合物中除去形成的氨.這一方法也適用于不同的二胺與氨基醇的反應.
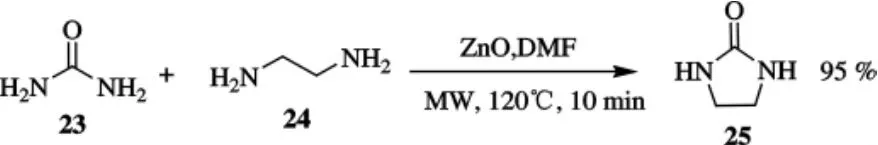
圖7 化合物25的合成路線
5 微波輔助合成苯并咪唑及其衍生物
通過鄰苯二胺與羧酸及其衍生物的反應合成苯并咪唑及其衍生物是一個相當傳統的合成路徑,傳統的方法往往需要較為苛刻的條件——強酸性條件、較高的溫度、很長的時間等.近年來,隨著微波技術在有機合成中的廣泛應用,極大的改善了用鄰苯二胺與羧酸合成苯并咪唑的條件.微波輻射鄰苯二胺與羧酸合成苯并咪唑具有速度快、產率高、污染少、安全性高等優點.
Vanelle等[25]研究發現,微波輻射可以有效地促進α-羥基羧酸與鄰苯二胺的反應合成2-羥甲基苯并咪唑,其反應效率較傳統加熱方法有明顯提高.傳統加熱合成時,需要120 h,而采用微波輻射時,反應僅僅需要1.5 h就能夠高收率合成2-羥甲基苯并咪唑26,合成路線如圖8所示.
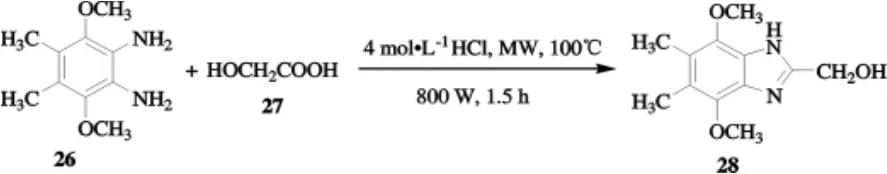
圖8 化合物28的合成路線
以鄰苯二胺和醛為原料合成苯并咪唑也是一個較為傳統的方法,這一傳統方法存在反應時間較長、副反應復雜、分離提純困難等缺點.將傳統的加熱方式改為微波輻射,將會更好地改善合成步驟、縮短反應時間、降低生產成本等.Jacob等[26]在微波輻射下,用 SiO2/ZnCl2作催化劑,合成了1,2-雙取代苯并咪唑31,合成路線如圖9所示.反應時間為1.5min,收率較高.其中,R為苯環時,用傳統的加熱方式,溫度為75℃,收率為65%,而利用微波輻射反應,溫度降低為65℃,收率可達92%.

圖9 化合物31的合成路線
6 結語
綜上所述,微波輻射合成咪唑類化合物較傳統的合成方法有以下優點:一是使咪唑的合成從傳統的污染性、有毒性逐漸走向“綠色合成”;二是極大地縮短了反應的時間,產率更高,反應外觀更加干凈;三是對于某個給定的咪唑合成反應,溶劑的選擇不再取決于沸點高低,而更傾向于反應介質的介電能力;四是由于微波輻射合成周期短,為未來咪唑藥物分子的篩選提供了廣闊的分子庫.因此,采用微波輻射技術,給咪唑的合成帶來了簡潔、方便的途徑,也為未來咪唑合成方法提供了光明的前景.我們相信,隨著微波技術的不斷發展,咪唑的微波合成將會得到快速發展,這將為尋找結構新穎、生物活性良好的咪唑類藥物中間體提供更加廣闊的選擇空間.
[1]C O卡帕,A斯塔德勒.微波在有機和醫藥化學中的應用[M].麻遠,譯.北京:化學工業出版社,2007:206-209.
[2]Yoon J,Kim S K,Singh N J,et al.Imidazolium receptors for the recognition of anions[J].Chem Soc Rev,2006,35(4):355-360.
[3]周成合,張飛飛,甘淋玲,等.超分子化學藥物研究[J].中國科學B輯:化學,2009,36(3):208-252.
[4]Zhou C H,Gan L L,Zhang Y Y,et al.Review on supermolecules as chemical drugs[J].Sci China Ser B:Chem,2009,52(4):415-458.
[5]Zhang F F,Gan L L,Zhou C H.Synthesis,antibacterial and antifungal activities of some carbazole derivatives[J].Bioorg Med Chem Lett,2010,20(6):1881-1884.
[6]Wang X L,Wan K,Zhou C H.Synthesis of novel sulfanilamide-derived 1,2,3-triazoles and their evaluation for antibacterial and antifungal activities[J].Eur J Med Chem,2010,45(10):4631-4639.
[7]孟江平,耿榮霞,周成合,等.苯并咪唑類藥物研究進展[J].中國新藥雜志,2009,18(16):1505-1514.
[8]袁勇,周成合,劉嬙,等.新型合成抗菌藥物研究新進展[J].中國新藥雜志,2007,16(5):343-349.
[9]Jiang H Y,Zhou C H,Luo K,et al.Chiral imidazole metalloenzyme models:Synthesis and enantioselective hydrolysis for α -amino acid esters[J].J Mo Cat A:Chem,2006,260(1-2):288-294.
[10]吳俊,米佳麗,周成合.組胺H3受體配體研究進展[J].中國藥學雜志,2007,42(6):404-409.
[11]蔡佳利,李碩,周成合,等.咪唑類抗癌藥物研究進展[J].中國新藥雜志,2009,18(7):21-31.
[12]孟江平,盧一卉,海力且木·依不那音,等.苯并咪唑類酶抑制劑研究進展[J].中國生化藥物雜志,2008,29(6),422-425.
[13]孟江平,于克貴,甘淋玲,等.大環類超分子藥物研究[J].西北師范大學學報:自然科學版,2008,44(suppl):242-245.
[14]孟江平,于克貴,周成合.2,4-二氯芐類苯并咪唑環番的合成[J].有機化學,2007,27(suppl):557.
[15]Zhou C H,Zhang Y Y,Yan C Y,et al.Recent researches in metal supramolecular complexes as anticancer agents[J].Anti-Cancer Agents in Med Chem,2010,10(5):371-359.
[16]Wolkenberg S E,Wisnoski D D,Lindsler W H,et al.Efficient synthesis of imidazoles from aldehydes and 1,2-diketones using Microwave irradiation [J].Org Lett,2004,6(9):1453-1456.
[17]Sparks R B,Combs A P.Microwave-assisted synthesis of 2,4,5-triaryl-imidazole;A Novel Thermally Induced N-Hydroxyimidazole N—O bond cleavage[J].Org.Lett,2004,6(14):2473-2475.
[18]朱映光,瞿昌偉,胡文浩.不對稱多組分反應[J].有機化學,2010,22(7):1380-1396.
[19]Sha F,Huang X.A multicomponent reaction of arynes,isocyanides,and terminal alkynes:Highly chemo-and regioselective synthesis of polysubstituted pyridines and isoquinolines[J].Angew Chem Int Ed,2009,48(19):3458-3461.
[20]陳育蘭,嚴勝驕,林軍.多組分反應在雜環合成中的應用[J].廣東化工,2010,37(3):4-6.
[21]Ireland S M,Tye H,Whittaker M.Microwave-assisted multi-component synthesis of fused 3-aminoimidazoles[J].Tetrahedron Lett,2003,44(23):4369-4371.
[22]Coleman C M,Mac Elroy J M D,Gallagher J F,et al.Microwave parallel library generation:Comparison of a conventional-and microwave-generated substituted 4(5)-sulfanyl-1H-imidazole library[J].J Comb Chem,2002,4(1):87-93.
[23]劉金艷,肖小明,譚年元,等.微波輻照2,6-二(2'-咪唑啉-2'-基)吡啶的合成及其與DNA的相互作用[J].應用化學,2010,27(6):658-663.
[24]Merriman G H,Ma L,Shum P,et al.Synthesis and SAR of novel 4,5 diarylimidazolines as potent P2X7receptor antagonists [J].Bioorg Med Chem Lett,2005,15(4):435-438.
[25]Boufatah N,Gellis A,Maldonado J,et al.Efficient microwave-assisted synthesis of new sulfonylbenzimidazole-4,7-diones:heterocyclic quinones with potential antitumor activity[J].Tetrahedron,2004,60(41):9131-9137.
[26]Jacob R G,Dutra L G,Radatz C S,et al.Synthesis of 1,2-disubstitued benzimidazoles using SiO2/ZnCl2[J].Tetrahedron Lett,2009,50(13):1495-1497.
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
