老年癡呆藥物研究進展
2011-05-17 09:55:14謝雨禮
藥學與臨床研究 2011年1期
關鍵詞:研究
謝雨禮
大冢(上海)藥物研究開發有限公司,上海 201203
癡呆病(Dementia)是一種持續性的智能障礙,病因十分復雜,既有遺傳因素,又有后天的環境影響和誘發,可引起癡呆的疾病高達百余種。其中,老年人常犯的癡呆主要有兩種:一是老年性癡呆,即阿爾茨海默氏病(Alzheimer’s disease, AD);二是血管性癡呆(Vascular Dementia,VD)。這兩種類型占到老年期癡呆的80%以上。老年癡呆病,由德國醫生Alois Alzheimer于1906年首先發現,因此以其名字命名。這是一種慢性進行性的神經退行性疾病,早期表現為短期記憶力缺失,隨著疾病的發展,大腦神經細胞功能逐漸喪失,造成記憶力、判斷力、方位感、注意力和語言能力的損傷,并伴有行為和人格方面的改變,最終在5~10年內導致病人死亡。近年來,AD的發病率逐年攀升,已經成為嚴重威脅老年人生命健康和生活質量的主要疾病之一。根據阿爾茨海默氏病國際(AD International)的年度報告[1],2010年全世界年齡大于60歲的老人達7.5854億,其中約4.7%、3556萬人患有不同程度的AD。到2030年和 2050年,這個數字將大幅上升,分別達到6569萬和11538萬,其中尤以中國和印度等發展中人口大國增加最為迅速。在我國,大多數人認為中國的AD發病率低于西方國家,其實這是個誤區,主要原因是人們一直認為記憶力衰退是人衰老的一部分,不是疾病,也很少到醫院就診。我國老年癡呆病的大規模調查表明,中國并非癡呆病的低發區,發病率與歐美發達國家相近,而且AD的發病率要明顯高于血管性癡呆[2]。由于AD病人生活不能自理,長期需要專人照顧,因此給病人帶來痛苦的同時,也給家庭和社會帶來了沉重的經濟和心理負擔,已經成為不可忽視的社會問題。瑞典的Karolinska研究所2005年的一項研究估計,全球癡呆病的花費,如果包括非直接的費用,每年高達3000多億美元,其中AD占到絕大部分[3]。AD非常頑固難治,其發病機制也不是很清晰,迄今尚未找到治愈該病的有效方法。目前AD一線治療藥物,比如乙酰膽堿酶(AChE)抑制劑,只能緩解早期病人的認知障礙,提供適度的癥狀改善作用,無法阻止病情的進展。因此尋找具有治療作用 (disease-modifying)的抗AD藥物成為當前藥物開發的熱點。由于AD帶來的社會問題,這一領域也已引起世界各國政府的高度重視,成為優先支持的研究之一。近年來,神經生理、生化和藥理等方面的基礎研究不斷深入,導致AD藥物的研究和開發不斷取得階段性進展,但大多集中在臨床前研究。最近幾個寄予厚望的新藥,如輝瑞的Latrepirdine和禮來的Semagacestat,三期臨床相繼失敗,讓人們重新開始審視AD藥物的研發策略,主要集中在對AD發病機制的理解,診斷的缺陷,靶點的選擇以及臨床方案的設計等方面。本文將對AD的發病機制作簡單介紹,并回顧基于這些機制的上市藥物和臨床研究藥物,希望從這些成功和不成功的案例中得到啟發,為今后AD藥物的開發和研究提供思路和方向。
1 AD的發病機制
如圖1所示,AD在病理方面具有兩大顯著特征:一是 β 淀粉樣多肽(β-amyloid Peptide, Aβ)在大腦皮層和海馬區神經細胞外累積沉淀形成β淀粉樣斑(Aβ plagque);二是腦神經細胞內Tau蛋白異常聚集形成的神經原纖維纏結(neurofibrillary tangles)。另外,神經元突觸功能異常,錐體神經細胞丟失以及乙酰膽堿等神經遞質的大量降解也是較為常見的病理改變。

自20世紀90年代提出所謂的 “Amyloid Hypothesis”以來,人們普遍認為Aβ是AD發病機制的核心。具有神經毒性的Aβ在中樞神經系統累積誘導神經細胞凋亡,引起和促進一系列病理變化,最終導致AD復雜多樣的病理特征和臨床表現。因此以抑制和清除Aβ或其聚集的斑塊為目的治療手段成為當前藥物開發的重要方向之一。盡管Aβ假說一直受到爭議,但目前還沒有其它理論能夠替代它當前在AD研究領域的地位。首先,Aβ沉積是老年癡呆最典型的病理特征之一,Aβ沉積和疾病的發生和進展具有很強的相關性,通過老年斑診斷老年癡呆,準確率達到80%以上,另外10%~20%誤診一般都屬于其它癡呆類型[4]。最近的研究表明,大腦區域的Aβ40和Aβ42含量幾乎與AD病人臨床上認知能力損傷具有定量的相關性,特別是在疾病的早期階段表現尤為突出[5]。另外,已經成熟的Aβ蛋白可能不是唯一的致病因素,可溶性的、體積較小的Aβ分子寡聚體神經毒性更強,與神經細胞的損傷具有密切的相關性[6]。第二個強有力支持Aβ假說的證據是基因突變方面的。變異的Aβ前體蛋白(APP)或參與其切割的蛋白,如Presenilin 1和2的突變都會異常增加Aβ的產生,從而導致早期發生的老年癡呆或家族性老年癡呆(familiar AD)。雖然家族性老年癡呆在所有病例中只占到2%左右,但這些基因突變攜帶者幾乎100%會患AD,強烈支持Aβ在AD發病機制中的關鍵作用[7]。大部分AD是所謂的零星發生的老年癡呆(Sporadic AD),一般發生較晚,遺傳性也沒有家族性AD這樣強,然而有關基因的變異也能大大增加患病的可能性。目前研究最多的是一個與脂質代謝有關的基因ApoE,雖然ApoE變異致病沒有象APP和Presenilin一樣與Aβ直接相關,其他功能可能也參與作用,但ApoE變異確實也能加快Aβ的沉積[8]。轉基因動物,特別是轉基因小鼠過度表達APP等與Aβ相關的人類基因,能夠重現AD的一些典型病理特征,而且表現出與年齡相關的學習和記憶力的衰退,成為研究AD和開發藥物的有用工具,也為Aβ假說提供了大量的動物實驗證據。然而,Aβ通過何種機制調節其細胞神經毒性還不是很清楚,有待進一步研究。
大部分基于Aβ假說的藥物還處于各個階段的研究當中,目前還沒有成功上市的例子。倒是幾個抑制Aβ產生的小分子藥物和清除Aβ斑塊的疫苗臨床研究失敗案例為Aβ假說的反對者提供了依據。其他與Aβ假說競爭的理論也因此日益受到重視。其中最具代表性就是以Tau為核心的致病機理。上述提到神經細胞里含有高度磷酸化Tau形成的神經原纖維纏結是老年癡呆另一個標志性病理特征。有研究表明,認知紊亂與纖維纏結的位置和含量有密切的相關性[9]。變異的Tau可導致前頭側頭型癡呆(frontotemporal dementia),這表明 Tau 與癡呆癥直接相關[10]。雖然這類癡呆在病理特征和臨床癥狀上與AD有所不同,但表明Tau異常可以引起神經細胞丟失和認知紊亂,在AD中絕不只是病變結果這么簡單。Tau是一種細胞質蛋白,與微管蛋白(Tubulin)有很高的親和性,可以穩定神經元微管結構,保證神經元運輸通道的暢通。在老年癡呆病人中,Tau被過度磷酸化,從而聚集成不溶的纖維纏結。這些纖維纏結有直接的神經毒性,也可消耗可溶性的Tau,阻斷神經元的運輸通道。兩種因素都可能導致疾病的發生和惡化[11]。
線粒體功能紊亂 (Mitochondrial dysfunction)在AD中出現較早,常常導致神經細胞損傷和凋亡,被認為是神經性退變的原因之一[12]。APP和Aβ通過特殊運輸機制,能夠進入線粒體中與許多線粒體關鍵元件相互作用,破壞能量代謝和其正常功能。大腦炎癥(Inflammation)是AD重要的臨床表現之一。臨床統計表明,服用非甾體抗炎藥可減少患老年癡呆的風險[13]。最近一項基因篩選發現了CR1(complement receptor 1),它是一個調節自身免疫的關鍵基因,其突變是患老年癡呆的危險因素之一,從而建立了炎癥和老年癡呆的基因鏈接[14]。其他AD的致病機制還包括活性氧化自由基(ROS)的大量產生對大腦分子的損傷;鈣、銅、鋅、鉀等陽離子代謝紊亂;脂質和能量代謝紊亂等[15]。基于這些機制開發的藥物,都已經進入臨床研究。
2 AD的治療藥物
隨著人口老齡化,老年癡呆的發病率迅速升高,其治療藥物市場前景巨大。據Research and Market的分析報告[16],2008年全球老年癡呆藥物市場達到54億美元,到2015年將會達到64億美元。輝瑞、禮來等世界制藥巨頭都在加快抗AD藥物的開發,以期在未來市場競爭中奪得先機。除上市藥物外,截至2010年,世界藥物研究開發信息庫“Pharmaprojects”中收錄處于臨床的AD藥物高達130余個,還有數百個處于臨床前研究。這些藥物包括小分子化合物、疫苗、抗體、特殊制劑,以及神經元再生和干細胞治療等多種治療手段。以下將對部分上市和處于臨床研究的AD藥物作一簡要介紹。
2.1 上市藥物
臨床上用于治療AD的藥物大概有10余種,主要是擬膽堿藥物和改善腦代謝的益智藥。現已知腦內乙酰膽堿的含量與記憶力密切相關。老年癡呆病人大腦中乙酰膽堿大量減少,引起記憶力衰退,補充膽堿類物質能改善其記憶力和思維能力。直接給予膽堿或卵磷脂,由于代謝太快,不能增加乙酰膽堿的含量。而抑制乙酰膽堿水解酶可延緩乙酰膽堿的代謝分解,從而延長突觸后受體的興奮,改善記憶力。乙酰膽堿酶抑制劑是目前治療AD的一線藥物。
如圖2所示,美國FDA批準的5個抗老年癡呆藥物,其中4個為乙酰膽堿酶抑制劑,包括他克林(Tacrine)、多奈派齊(Donepezil)、利斯的明(卡巴拉汀,Rivastigmine)和加蘭他敏(Galanthamine)。 國內研制的哈伯英或石杉堿甲,是我國學者從石杉屬植物千層塔分離得到的一種生物堿,也是主要通過抑制乙酰膽堿酶達到改善記憶的療效。乙酰膽堿酶抑制劑只能改善AD病人的癥狀,而不能延緩疾病的進程,主要對于早期AD病人療效較好。鹽酸美金剛胺是唯一用于中度和嚴重階段AD的藥物。美金剛胺的作用機制與乙酰膽堿酸抑制劑也有所不同,主要是通過阻斷NMDA受體,延緩能損害神經細胞的興奮神經遞質谷氨酸(Glutamate)的釋放。由于作用機制不同,美金剛胺可與乙酰膽堿酶抑制劑合用,增加療效。比如,在一項由404位中度和嚴重的AD病人參加的三期臨床研究中,美金剛胺和多奈派齊合用比單獨使用多奈派齊取得明顯更好的療效[17]。這些藥物除他克林由于肝臟毒性逐漸退出市場外,在國內都有銷售。如圖3所示,輝瑞的多奈哌齊目前在國內市場上處于領先地位,占到市場份額的58%左右。石杉堿甲和Forest Laboratories的美金剛胺市場份額相當,但美金剛胺應該更具前景。
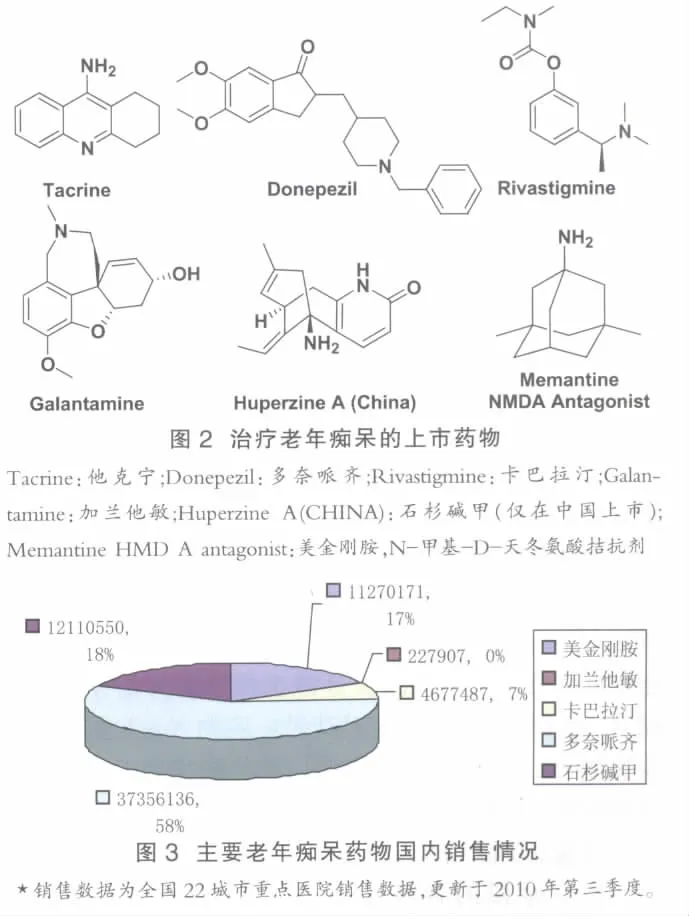
改善腦代謝的益智藥一般具有擴張腦血管的作用,臨床上常用于腦損傷后的恢復,這些藥物對老年癡呆的某些癥狀如記憶力衰退、適應環境能力降低等有不同程度的改善作用。常用于老年癡呆的藥物包括西坦類藥物,吡硫醇,鈣離子拮抗劑尼莫地平等。
2.2 處于臨床研究的藥物
目前上市的藥物對老年癡呆都只能起到改善癥狀的作用。因此到目前為止,仍缺乏控制疾病進程的有效治療手段。基于Aβ假說和Tau蛋白等新機制開發的藥物都還處于臨床研究階段。
2.2.1 以Aβ為靶點的藥物
2.1.1.1 降低Aβ產生的藥物
APP代謝產生Aβ的過程是APP被β和γ分泌酶 (beta-and gamma-secretase)裂解為具有40、42或43個氨基酸的Aβ小肽。Aβ分子具有自發聚合的能力,它能在細胞外聚集成寡聚體、纖維和斑塊等各種存在形式。因此抑制β和γ分泌酶成為阻止Aβ分子產生的有效手段(表1)。β分泌酶有許多底物,因此含有廣泛的底物結合區域,另外抑制劑還需通過血腦屏障,開發其抑制劑很具有挑戰性。目前只有5個化合物進入了一期臨床。其中CoMentis開發的CTS-21166在健康成年人中口服給藥,可以降低血漿中Aβ的含量,并且具有很好的耐受性[18]。有意思的是降血糖藥物羅格列酮(Rosiglitazone)和吡格列酮 (Pioglitazone),通過刺激PPARγ受體可以抑制β分泌酶的表達,并促進APP的降解,從而減少Aβ的產生[19]。然而,臨床研究表明,這些藥物對AD病人的認知能力并沒有改善作用[20],加之最近FDA對羅格列酮潛在心臟毒性的警告,PPARγ激動劑作為AD治療藥物的開發都已停止。
γ分泌酶負責Aβ產生的最后一步裂解,其抑制劑開發曾經是AD藥物研發的熱點之一。禮來的小分子γ分泌酶抑制劑Semagacestat(LY-450139)能夠有效地減少Aβ的產生,進入到了三期臨床研究。令人失望的是Semagacestat不但沒有治療作用,還加快病人認知功能的喪失,因此禮來于 2010年9月份宣布終止其臨床研究[21]。另外幾個藥物,如Merck的MK-0752,輝瑞的 PF-3084014 和 GSI-9531(begacestat)也已終止臨床研究。目前還有5個γ分泌酶抑制劑處于一期和二期臨床研究。其中Mount Sinai醫學院開發的NIC5-15是一個天然的單糖化合物,除增加胰島素的靈敏性外,還能選擇性的抑制γ分泌酶對Aβ的裂解作用,而對其另一底物Notch蛋白卻沒影響[22]。二期臨床研究表明該藥物治療老年癡呆安全有效[23]。另外,部分非甾體抗炎藥被發現可以調節γ分泌酶,抑制Aβ1-40和Aβ1-42的產生,而升高無害的Aβ1-38片段的含量。這些化合物中,Encore的Tarenflurbil(R-enantiomer of Flurbiprofen)進入到三期臨床,但未能顯示好的臨床結果[24]。
有研究表明,升高alpha-分泌酶的活性,可以增加APP的良性代謝而降低Aβ的形成,而且其代謝產生的可溶性片段sAPP-alpha還有神經保護作用[25]。法國公司ExonHit開發的Etazolate是一種口服小分子藥物,選擇性調節GABA受體并可抑制PDE4,能夠刺激alpha-分泌酶的活性,升高sAPP-alpha的產生。目前,該藥物二期臨床研究結束,但結果沒有披露。美國Aphios開發的抗癌藥物Bryostatin-1通過激活磷酸激酶PKC刺激alpha-分泌酶,促進sAPP-alpha的分泌,在老年癡呆動物模型中顯示了較好的活性[26],該藥物的二期臨床研究正在計劃當中。美國ProteoTech和我國的天士力公司共同開發的口服小分子抗老年癡呆藥物Exebry-1在美國已經進入一期臨床研究。該藥物具有多種活性,除作用于Tau蛋白和抗炎外,也能調節beta和alpha分泌酶的活性,抑制Aβ的產生[27]。
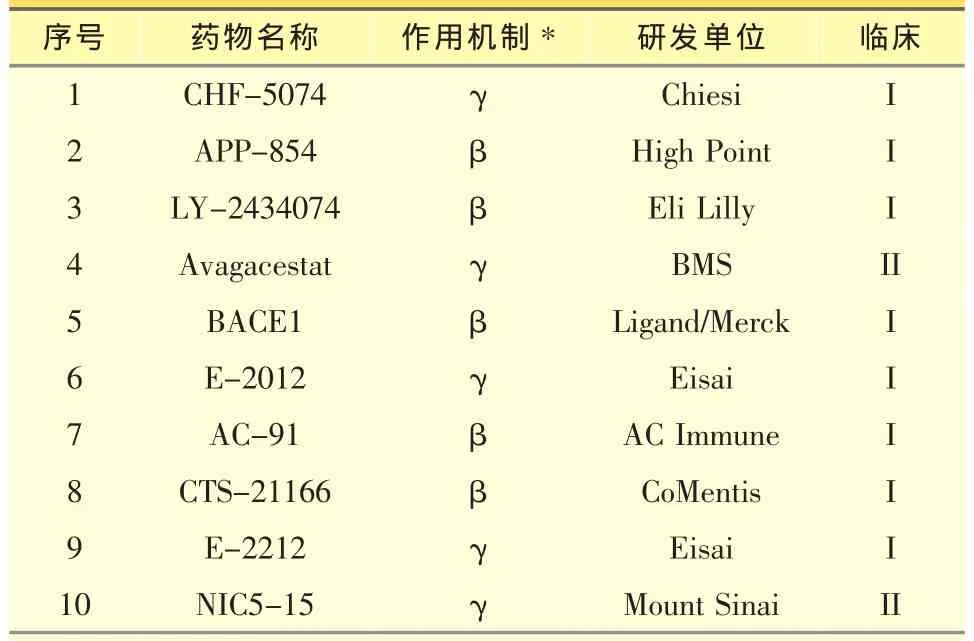
表1 處于臨床研究的β和γ分泌酶抑制劑
2.2.1.2阻止Aβ聚合的藥物
阻止Aβ分子聚合成具有神經毒性的寡聚體或斑塊是基于Aβ假說開發AD藥物的方向之一。抗聚合的化合物一般通過與Aβ單體分子結合,阻止其聚合和加快清除代謝。Bellus Health開發的tramiprosate是第一代的抗聚合藥物。本品是口服有效的小分子化合物,傾向于與可溶性的Aβ分子結合而維持其非聚合狀態,可惜該藥物三期臨床結果也不理想[28]。Bellus后于2008年將其開發成OTC營養品上市銷售(商標名Vivimind)。 NRM-8499是該公司開發的tramiprosate前藥,已進入一期臨床。金屬離子螯合劑可以與促進Aβ聚合的銅離子和鋅離子結合,從而抑制Aβ聚合物的形成。Clioquinol(PBT1)是第一個進入臨床研究的抗AD螯合劑,由于合成工藝問題,Prana Biotech放棄了該藥物的開發,而將活性更好的類似物PBT2推上了二期臨床。在一項為期12周的二期臨床試驗中,PBT2降低了大腦CSF中的Aβ1-42的含量,并改善了病人整體功能,本品的耐受性也非常好[29],不知什么原因該藥物的三期臨床研究一直沒有啟動。AZD-103是愛爾蘭公司Elan開發的可透過血腦屏障的肌醇立體異構體,它可以與Aβ結合,調節Aβ分子的折疊,促進Aβ聚合物的分解。臨床研究表明,本品高劑量(>1000mg·d-1)時有較大副作用,其低劑量(250mg·d-1)組的二期臨床仍在進行中[30]。綠茶中得到的多酚化合物EGCg具有多種治療AD的活性,其中之一就是抑制Aβ的聚合,其對早期AD病人的二期臨床試驗還在進行當中(NCT00951834)。
2.2.1.3 加速Aβ清除的藥物
通過免疫的手段來清除Aβ是AD藥物研發領域的重大突破之一。使用疫苗和抗體主被動免疫的策略,至少可通過三種機制清除Aβ:與Aβ結合,加速其聚合物的溶解;巨噬細胞的吞噬以及抗體的“提取”作用。Elan的科學家首次發現抗Aβ疫苗在動物實驗中對AD有很好的治療作用[31],隨后該公司的Aβ疫苗AN-1792在二期臨床研究中失敗,主要是一部份病人發生了嚴重的腦部炎癥。而且隨后發現,雖然AN-1792降低Aβ的功能強大,甚至能完全清除病人腦中的Aβ老年斑,對病人認知功能卻沒有改善作用[32]。諾華的CAD-106,目前處于二期臨床,能誘導特異的抗Aβ抗體但不引起CNS炎癥,前景較為看好[33]。其他還有4個類似的疫苗處于早期臨床階段(表2)。值得一提的是,GSK和AFFiRis共同開發的AD-02是模仿天然Aβ的人工合成的小肽,能夠誘導抗體特異性進攻Aβ的N端片斷,在動物模型中療效較好,目前已進入二期臨床研究[34]。
被動免疫的策略是直接用抗體進攻Aβ分子,這種方法可以允許發展特異性更強的抗體,而且在老年病人中,這種方法可能更有效,因為老年人對疫苗的免疫反應能力普遍下降。目前至少有9個人源化的單克隆抗體處于臨床研究,其中輝瑞的Bapineuzumab和禮來的Solanezumab已進入三期。Bapineuzumab能夠與Aβ的N端片斷緊密結合,主要進攻Aβ斑塊,兩項二期臨床研究顯示它能有效降低Aβ的含量,但對認知能力的改善沒有統計學差異,似乎對不含 ApoEε4等位基因(ε4 allele)的個體更有效[35]。有意思的是,本品還可以降低CSF中Tau的含量[36]。該藥物最大的副作用是血管源性腦水腫,幾乎10%的臨床參與者受到影響,而且副作用在攜帶有ApoEε4等位基因的個體中更為明顯,不幸的是ApoEε4等位基因正是患老年癡呆的危險因素之一。目前,Bapineuzumab仍在進行三期臨床研究[37]。禮來的Solanezumab主要作用于可溶性的Aβ分子,可加速Aβ的清除,而且能進攻幾個Bapineuzumab無效的Aβ變種分子,本品能有效的降低腦中Aβ的含量[38],兩項三期臨床研究已經展開,將進一步研究其對早中期AD的療效。另外,輝瑞的Ponezumab和羅氏的Gantenerumab也已進入二期臨床研究,其它5個抗Aβ抗體還在一期進行安全性評價(表2)。

表2 處于臨床研究的抗Aβ的疫苗和抗體
除生物制品外,也有少數小分子化合物被發現,可加速Aβ的清除。美國Archer公司開發的鈣離子拮抗劑Nilvadipine和其類似物ARC-031就是例子之一,這些藥物都還處在一期臨床,療效有待觀察。
2.2.2 以Tau蛋白為靶點的藥物Tau被認為在老年癡呆的致病機理中扮演重要的角色,因此也成為抗AD藥物重要的靶點之一。目前主要有兩種方法靶向Tau:一是抑制Tau的磷酸化,磷酸激酶調節的過度磷酸化是Tau異常聚合的主要原因;二是直接抑制Tau的聚合或促進其聚合物的分解。GSK-3是調節Tau磷酸化最主要的激酶。精神病治療藥物Valproate和鋰被發現可抑制GSK-3,降低Tau的磷酸化程度,具有神經保護作用,可惜臨床研究發現這兩個藥物對AD病人的認知能力和整體功能沒有改善作用[39]。Noscira開發的非ATP競爭性GSK-3抑制劑Tideglusib目前仍在進行二期臨床研究,還沒有公開任何研究結果。亞甲基藍是一種常用的染料,具有抗自由基的活性,并能直接抑制Tau的聚合,在動物實驗中,單獨或與乙酰膽堿酶抑制劑卡巴拉汀合用可扭轉記憶力缺失和學習能力損傷[40]。臨床研究表明,亞甲基藍在60 mg劑量時可改善中期AD病人的認知功能,但100 mg組沒有效果,被認為是劑型的缺陷造成的[41],TauRx公司最近又開發了其新的劑型TRx0037,已進入一期臨床研究。Allon Therapeutics最近基于神經保護蛋白ADNP開發了一個八肽化合物Davunetide可調節微觀蛋白,抑制Tau的磷酸化和聚合,二期臨床試驗表明,其對中等嚴重的AD病人有較好的療效[42]。煙酰胺是維生素B3的生物活性體,研究顯示該化合物可穩定微觀結構,降低磷酸化Tau的含量,改善AD動物的記憶力[43]。
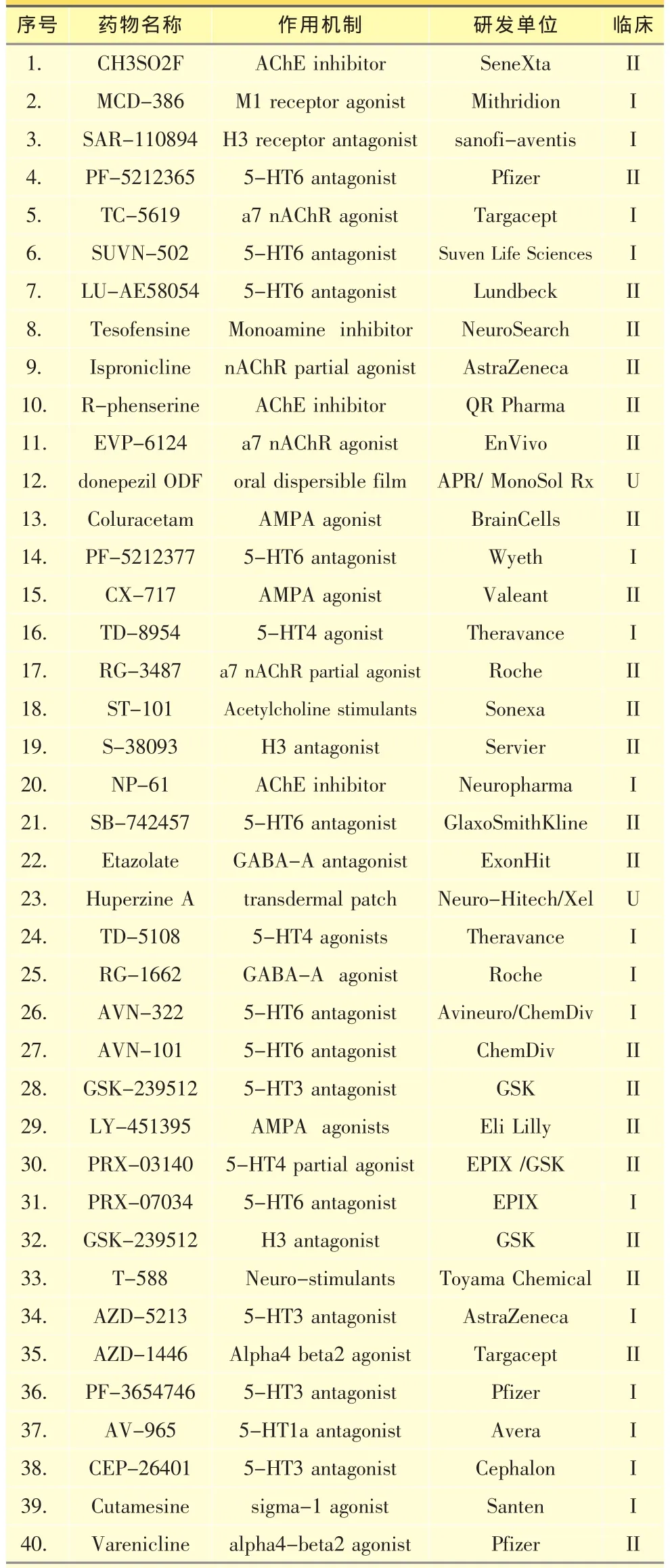
表3 處于臨床研究的基于神經遞質的藥物
2.2.3 基于神經遞質的藥物神經遞質紊亂與AD病理特征和記憶力衰退等臨床表現直接相關。比如乙酰膽堿酶抑制劑就是基于AD中膽堿通路的嚴重缺失而發展的。這些藥物依靠膽堿神經末梢起效,隨著疾病的發展,這些末梢不斷退化,因而此類藥物也慢慢失去效果。神經遞質乙酰膽堿參與的各種活動主要通過突觸后膜兩大受體系統發揮作用,即毒蕈堿型受體(M受體)和煙堿型受體(N受體)。發展這些受體的小分子激動劑成為研發AD藥物的新方向。另外,5-羥色胺、組胺、AMPA、GABA和NMDA受體調節劑也日益受到重視。調節這些受體不但能改善認知能力,也可能降低Aβ和Tau起到治療疾病的作用[44]。表3列出40個目前處于臨床研究的基于神經遞質的藥物,其中包括石杉堿甲和多奈派奇等上市藥物的新制劑,這些藥物大多還處于一二期早期研究階段,由于篇幅有限,本文不做詳細介紹。
2.2.4 抗炎、抗氧化劑等其他作用機制的藥物大量研究表明,抗炎和抗氧化劑對AD具有一定的治療作用。非甾體抗炎藥物,比如輝瑞的COX-2抑制劑Celecoxib用于老年癡呆的治療,曾進入三期臨床研究,但后來由于結果不理想被終止了。表4列出一些仍在進行臨床研究的抗炎和抗氧化劑以及一些新作用機制藥物 (如HDAC和PDE抑制劑等)。其中值得注意的是干細胞治療 (HuCNS-SC,Stem-Cells)、細胞治療(ECB-AD,NsGene A/S)和基因治療(CERE-110,Ceregene)等新的治療手段已經進入早期臨床研究階段,可能給AD的治療帶來新的希望。另外,幾個具有神經保護等多種藥理活性的植物提取物(INM-176,SK-PC-B70M,Anapsos)也在后期臨床研究階段,其治療效果,令人拭目以待。

表4 處于臨床研究的抗炎,抗氧化劑等其他作用機制的藥物
結 語
隨著基礎研究日新月異,人們對AD的致病原因理解越來越深入。比如,Science最近報道一項研究結果表明,AD病人腦中Aβ分子的產生并沒有受到影響,而是其清除受到抑制,并且發生遠遠早于疾病的臨床表現[45]。因此,抑制Aβ分子產生的治療方法需要重新考慮。這也表明,早期診斷對于及時治療老年癡呆至關重要,等到疾病后期,大面積神經衰退可能難以逆轉。另外,具有多種作用機制的藥物,通過協同作用,可能比基于單個靶點或單種作用機制的藥物更具前景。雖然,輝瑞的Dimebon(Latrepirdine)最近臨床失敗,但該藥物通過多種機制,調節線粒體的功能而發揮治療作用,這種靶向整個器官的新思路應該受到重視。特別是我國中藥在這方面有一定的優勢,如果能夠合理設計臨床方案,科學評價藥物的療效,中藥在攻克AD的戰役中可能會成為一支“奇兵”。
[1] Alzheimer’s disease international,World Alzheimer Report,2009,Executive Summary.
[2]Zhang ZX,Zahner GE,Roman GC,et al.Sociodemographic variation of dementia subtypes in China:Methodology and results of a prevalence study in Beijing,Chengdu,Shanghai,and Xian.Neuroepidemiology,2006,27(3):177.
[3] Wimo A,Winblad B,Jonsson L.An estimate of the totalworldwide societalcostsofdementia in 2005.Alzheimer’s and Dementia,2007,7:13.
[4] Newell KL,Hyman,BT,Growdon ET,et al.J.Neuropathol Exp Neurol,1999,58:1147.
[5] Lue LF,Kuo YM,Rocher AE,et al.Am J Pathol,1999,155:853.
[6] Lesne S,Koh MT,Kotilinek L,et al.Nature,2006,440:352.
[7]Hardy J,Orr H.J Neurochem,2006,97:1609.
[8] Cherbuin N,Leach LS,Christensen H,et al.Cognit Disord,2007,24:348.
[9] Thal D.Exp Neurol,2000,163:98.
[10] Hutton M,et al.Nature,1998,393:702.
[11]GoedertM,KlugA,CrowtherRJ.AlzheimersDis,2006,9:195.
[12]Reddy PH,Beal MF.Trends Mol Med,2008,14:45.
[13]McGeer PL,McGeer E.Neurobiol Aging,2007,28:639.
[14] Lambert JC,et al.Nature Genet,2009,41:1094.
[15] Citron M.Nature Reviews,2010,9:387.
[16] Medical news,http://www.news-medical.net/news/20091209/Key-trends-impacting-the- global-Alzheimers-dis ease-market.aspx.
[17]Forest Laboratories Inc,Medical release,http://www.frx.com.
[18]Tang JJN.Alzheiers Dement,2009,5:74.
[19] Landreth G,Jiang Q,Mandrekar S.Neurotherapeutics,2008,5:481.
[20] Gold M,Alderton C,Zvartau ME,et al.Alzheimers Dement,2009,5:86.
[21]Lilly press release,http://news-room.lilly.com,Sep 18,2010.
[22]WangJ,HoL,PassinettiGM.AlzheimersDement,2005,1:62.
[23] Grossman H,Marzloff G,Luo X,et al.Alzheimers Dement,2009,5:259.
[24] Green RC,Schneider LS,Amato DA,et al.JAMA,2009,302:2557.
[25]Van Marum RJ.Fundam Clin Pharmacol,2008,22:265.
[26] Etcheberrigaray R,Tan M,Dewachter I,et al.Pro Natl Acad Sci USA,2004,101:11141.
[27] Snow AD,Cummings J,Lake T,et al.Alzheimers Dement,2009,5:418.
[28] Aisen PS,Saumier D,Ferris S,et al.61st American A-cademy of Neurology annual meeting,Seattle,WA,USA.
[29] Lannfelt L,Blennow K,Zetterberg H,et al.Lancet Neurol,2008,7:779.
[30] Press release,Dec 15,2009,Elan.http://newsroom.elan.com.
[31]Schenk D,Barbour R,Dunn W,et al.Nature,1999,400:173.
[32] Holmes C,Boche D,Wilkinson D,et al.Lancet,2008,372:216.
[33] Winblad BG,Minthon L,Floesser A,et al.Alzheimer Dement,2009,5:113.
[34] Schneeberger A,Mandler M,Otawa O,et al.J Nutr Health Aging,2009,13:264.
[35] RinneJO,BrooksDJ,RossorMN,etal.Lancet Neurol,2010,9:363.
[36] Blennow K,Zetterberg H,Wei J,et al.Alzheimer Dement,2010,6:s134.
[37] Kerchner GA,Boxer Al.Expert Opin Biol Ther,2010,10(7):1121.
[38] SiemersER,Friedrich S,Dean RA,etal.Clin Neuropharmacol,2010,33(2):67.
[39] Tariot PN,Aisen PS.J Clin Psychiatry,2009,70:919.
[40] Deiana S,Harrington CR,Wischik CM,etal.Psychopharmacology,2009,202:53.
[41] Gura T.Nat Med,2008,14:894.
[42] Schmechel DI, Gerard G, Vatakis NG, et al.Alzheimers Dement,2008,4:T483.
[43] Green KN,Steffan JS.Martinez-Coria H,et al.J Neurosci,2008,28:11500.
[44] Fisher A.Neurotherapeutics,2008,5(3):433.
[45] Mauuenyega KG,Sigurdson W,Ovod V,etal.Science,2010,330:1774.
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車工程學報(2017年2期)2017-07-05 08:13:02
國際商務財會(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
