Losartan對高氧致慢性肺疾病新生大鼠肺纖維化的干預作用
2010-08-21 13:46:26陳寧張丹單麗沈劉雪雁尚云曉薛辛東
中國醫科大學學報 2010年8期
關鍵詞:肺纖維化
陳寧,張丹,單麗沈,劉雪雁,尚云曉,薛辛東
(中國醫科大學 附屬盛京醫院小兒呼吸內科,沈陽 110001)
新生兒慢性肺疾病(chronic lung disease,CLD)作為早產兒肺透明膜病治療的并發癥在1967年首次被Northway描述,它也被稱為支氣管肺發育不良,以肺泡發育受阻、肺間質廣泛纖維化為主要病理特征。肺纖維化的發生是由許多細胞因子、炎性介質、內皮細胞和上皮細胞等構成的復雜的網絡。有研究表明,肺組織局部的腎素-血管緊張素系統可能是纖維化的發生中心環節,血管緊張素Ⅱ(angiotensinⅡ,AngⅡ)通過與血管緊張素Ⅱ1型受體(angiotensin Ⅱ type 1 receptor,AT1R)結合,調節細胞因子、細胞外基質沉積[1,2]并誘導肺泡上皮細胞凋亡[3],從而破壞正常的肺泡結構和功能,促進肺纖維化的發生。AT1R拮抗劑losartan可減輕博萊霉素誘導的纖維化動物模型的肺組織膠原的沉積[4,5]。近年研究表明,腎素-血管緊張素系統可能參與CLD 的發病過程[6,7],特異性 AT1R 拮抗劑 losartan可明顯降低高氧致CLD新生大鼠肺組織羥脯氨酸的表達[8]。本研究進一步觀察AT1R拮抗劑losartan對高氧致新生大鼠CLD肺組織膠原表達的影響,從而探討高氧致CLD可能的發病機制。
1 材料與方法
1.1 材料
動物模型的制備:將Wistar新生大鼠160只(中國醫科大學附屬盛京醫院實驗動物中心提供)出生后(連同母鼠)24 h內隨機分為4組:空氣組(組Ⅰ)、高氧組(組Ⅱ)、高氧+注射用水組(組Ⅲ)和高氧+losartan組(組Ⅳ)。組Ⅰ放置于空氣環境(FiO2為21%)中飼養。組Ⅱ~組Ⅳ置于氧箱中,持續輸入氧氣,維持氧濃度85%~90%,每日用測氧儀監測(FiCO2<0.5%,鈉石灰吸收CO2)。每日定時開箱0.5 h稱重、添水和飼料及更換墊料,并與組Ⅰ交換母鼠以免因氧中毒而導致代母鼠喂養能力下降。各組飼養條件均為室溫22~25℃,濕度60%~70%。在生后第6日開始,組Ⅳ每日將losartan(5 mg/kg)加到注射用水中按3 ml/kg灌胃,組Ⅲ每日給等量注射用水灌胃,至實驗結束。
1.2 方法
各組在實驗后的1、3、7、14和21 d腹腔內注射5%水合氯醛(6 ml/kg)麻醉后處死。立即打開胸腔,無菌條件下分離肺組織,取右肺各葉用冷生理鹽水洗凈殘血,吸干水分,置于無RNase的Eppendorf管中于-80℃冰箱中保存;取左肺置于4%多聚甲醛固定,常規脫水、石蠟包埋,制成蠟塊,待測。
1.2.1 HE染色光鏡觀察:每組動物各時間點隨機抽取8張切片,每張切片光鏡下(×200)隨機選取5個視野觀察病理改變。
1.2.2 Masson三聯染色光鏡觀察:光鏡下觀察膠原纖維呈藍色,胞漿呈紅色,胞核呈黑藍色。光鏡下觀察對比藍色膠原纖維所占比例和分布情況。
1.2.3 逆轉錄聚合酶鏈反應(reverse transcription polymerase chain reaction,RT-PCR)半定量方法檢測肺組織Ⅰ型膠原(collagen typeⅠ,ColⅠ)mRNA的表達:按說明書操作提取總RNA。半定量分析ColⅠmRNA的含量。擴增條件:94℃3 min,94℃40 s,52 ℃ 1 min,72 ℃ 90 s,35個循環后 72 ℃ 7 min;擴增產物201 bp。引物序列由上海博亞生物技術有限公司合成,上游:5′-TTCAGTGGCTCGCTTCGG-3′;下游:5′-TCCAGCACCAATCCCTTC-3′。
1.2.4 ELISA方法測定肺組織ColⅠ蛋白的含量:每組動物各時間點隨機抽取6份標本,稱50 mg濕重肺組織剪碎,冰盒內超聲粉碎、制成10%組織勻漿,按說明操作,結果以每mg肺組織所含ColⅠ的含量表示(ng/mg)。ELISA試劑盒購自上海森雄科技有限公司,考馬斯亮蘭蛋白測定試劑盒購自南京建成生物工程研究所。
1.3 統計學分析
數據以±s表示。應用SSPS11.5統計軟件進行統計學處理,多組間比較采用F檢驗,兩樣本均數間比較采用t檢驗。P<0.05為差異有統計學意義。
2 結果
2.1 光鏡觀察肺組織形態學改變
組Ⅰ7 d肺泡形態較規則,大小均勻,肺泡間隔稍厚;14和21 d肺組織進一步肺泡化,肺泡間隔變薄、肺泡隔增多、肺泡數量增多,21 d更為明顯。組Ⅱ7 d肺泡隔及肺泡腔內見巨噬細胞、中性粒細胞等炎性細胞浸潤,肺泡內出血,偶見肺不張,肺泡間隔增厚不明顯,終末氣腔擴張;14 d肺泡間隔較同期組Ⅰ增寬,肺間質細胞增多,肺泡數目減少,終末氣腔擴張明顯,偶見小灶肺實變;到21 d肺組織正常形態破壞,肺泡隔顯著增厚,肺泡數目明顯減少、結構簡單,終末氣腔擴張更為明顯、部分肺泡萎陷,并且肺出血及肺實變多見。組Ⅲ與組Ⅱ改變相似。組Ⅳ在7 d與組Ⅱ區別不明顯,14和21 d肺組織肺泡隔變薄,纖維化明顯減輕,但肺泡腔沒有明顯縮小。見圖1。
2.2 Masson染色觀察膠原沉積的變化
藍色的膠原纖維主要在支氣管及血管壁的外周,肺泡壁有少許。組Ⅱ隨高氧時間的延長,膠原纖維在支氣管及血管壁的外周沉積增加;肺泡間質沉積在7 d后逐漸增加,在肺泡周圍呈條索樣增生,14和21 d同組Ⅰ比較藍色膠原纖維所占比例明顯增加。組Ⅳ藍色膠原纖維所占比例較組Ⅱ減少且排列比較疏松,但仍明顯高于組Ⅰ。見圖2。
2.3 肺組織ColⅠ表達的動態變化
2.3.1 各組肺組織ColⅠ蛋白表達的變化:組Ⅱ、組Ⅲ和組ⅣColⅠ的含量均隨日齡增加逐漸增加(P<0.01)。7 d時組Ⅱ、組Ⅲ和組Ⅳ肺組織中ColⅠ蛋白的含量略高于組Ⅰ(P>0.05),14和21 d時較組Ⅰ顯著上升(P<0.01)。組Ⅳ在14 d時較組Ⅱ略有下降(P>0.05),21d時則顯著下降(P<0.01)。見表1。

表1 各組肺組織ColⅠ蛋白含量的變化(±s,ng/mg,n=6)Tab.1 Changes of ColⅠ protein in lung tissue of each group(±s,ng/mg,n=6)

表1 各組肺組織ColⅠ蛋白含量的變化(±s,ng/mg,n=6)Tab.1 Changes of ColⅠ protein in lung tissue of each group(±s,ng/mg,n=6)
1)P<0.01 vs groupⅠ;2)P< 0.01 vs groupⅡ.
Group 1 d after birth 3 d after birth 7 d after birth 14 d after birth 21 d after birthⅠ 473.78±27.78 455.67±49.88 438.44±51.27 430.59±36.20 431.80±28.11Ⅱ 457.73±37.30 468.99±29.21 486.69±30.47 521.99±37.141) 746.99±74.831)Ⅲ 457.73±37.30 468.99±29.21 471.09±49.91 534.00±43.041) 720.46±64.931)Ⅳ 457.73±37.30 468.99±29.21 483.89±48.23 502.19±43.411) 600.69±49.441),2)
2.3.2 各組肺組織ColⅠmRNA表達的變化:RTPCR結果顯示,組Ⅰ各時間點ColⅠmRNA表達無明顯差異(P>0.05),組Ⅱ和組ⅢColⅠmRNA表達隨日齡的增加顯著上升。7、14和21 d時組Ⅱ、組Ⅲ和組Ⅳ較組Ⅰ明顯上升(P<0.05或0.01)。組Ⅳ在14 d時較組Ⅱ略有下降,但差異不明顯,21 d時則顯著下降(P<0.01)。見表2,圖3。
表2 各組肺組織ColⅠmRNA表達相對量的比較(±s,n=8)Tab.2 Comparison with the expression of ColⅠ mRNA in lung tissue of each group(±s,n=8)
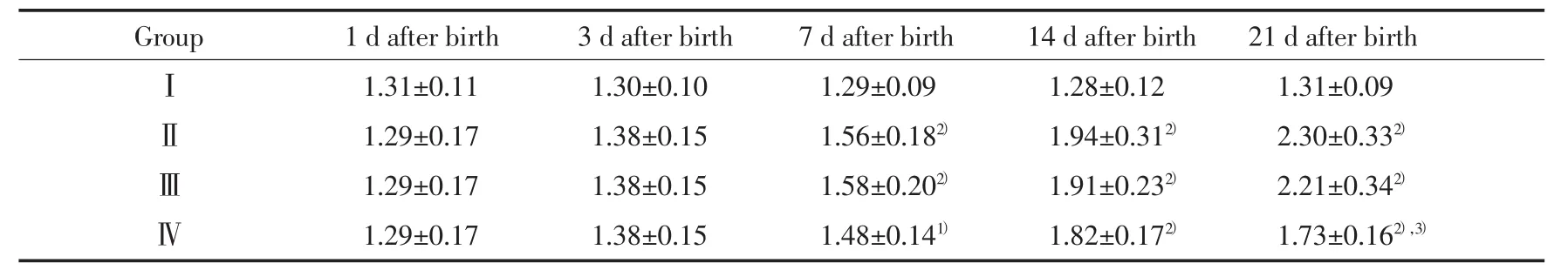
表2 各組肺組織ColⅠmRNA表達相對量的比較(±s,n=8)Tab.2 Comparison with the expression of ColⅠ mRNA in lung tissue of each group(±s,n=8)
1)P < 0.05,2)P < 0.01 vs groupⅠ;3)P < 0.01 vs groupⅡ.
Group 1 d after birth 3 d after birth 7 d after birth 14 d after birth 21 d after birthⅠ1.31±0.11 1.30±0.10 1.29±0.09 1.28±0.12 1.31±0.09Ⅱ1.29±0.17 1.38±0.15 1.56±0.182) 1.94±0.312) 2.30±0.332)Ⅲ1.29±0.17 1.38±0.15 1.58±0.202) 1.91±0.232) 2.21±0.342)Ⅳ1.29±0.17 1.38±0.15 1.48±0.141) 1.82±0.172) 1.73±0.162),3)
3 討論
肺內細胞外基質成分主要包括膠原蛋白、彈性蛋白、蛋白聚糖及糖蛋白4種。細胞與細胞外基質蛋白之間復雜相互作用對于肺的細胞生長、分支發育及肺的成熟具有重要作用。膠原蛋白是肺組織主要的細胞外基質,約占肺臟干重的五分之一。肺內主要有6種膠原類型,其中ColⅠ和ColⅢ分布于肺間質,是主要的結構蛋白。正常肺組織發育過程中,適量的膠原的合成有助于組織細胞的分化成熟、毛細血管的正常排列,對于肺結構和功能的建立有重要的作用。在病理情況下膠原也參與創傷修復和器官纖維化的形成過程,是肺損傷后修復、呼吸道結構重建所必需的。

過去曾認為肺間質纖維化的發生是由于病變肺組織存在持續的肺泡炎,集中在炎性因子的研究,而沒有考慮纖維化發生的初始因素。近年來研究認為上皮細胞損傷,上皮下成纖維細胞激活表型轉化為成肌纖維細胞,導致細胞外基質異常沉積是肺纖維化的原因所在。而這些微環境損傷修復的細胞事件伴隨著分子事件,即AngⅡ的產生。已經發現,在成人特發性纖維化、成人急性呼吸窘迫綜合征、硅肺等纖維化肺疾病中以及纖維化動物模型中,肺組織中AngⅡ和血管緊張素轉化酶濃度增加[9];并且AngⅡ以劑量依賴的方式促進成纖維細胞增殖。AngⅡ與成纖維細胞表面的AT1R結合,激活G蛋白,通過蛋白激酶C的級聯放大激活絲裂原激活蛋白激酶,促進成纖維細胞增殖和分化,同時誘導成纖維細胞自分泌轉化生長因子β[10]。應用AT1R拮抗劑losartan能夠顯著抑制AngⅡ誘導成纖維細胞的增殖與分化,抑制纖維化肺組織轉化生長因子β表達及膠原沉積[1]、延緩博萊霉素誘導大鼠肺纖維化的進程[11]。這些提示AngⅡ在組織損傷、纖維化過程中可能扮演較為重要的角色。然而Keogh[12]同樣在博萊霉素誘導肺纖維化實驗中卻得出相反的結論,認為肺纖維化疾病的發生是不依賴AngⅡ的誘導發生的,肺纖維化不能依靠losartan作為治療策略。Chen[13]在研究百草枯誘導的肺纖維化時發現,纖維化肺組織中轉化生長因子β及膠原含量增加與肺局部腎素-血管緊張系統無關。造成分歧的原因可能與不同品系的動物遺傳背景不同、纖維化發生的誘因不同以及藥物的劑量等因素所致。
近來研究發現在高氧CLD發病過程中也伴有肺局部腎素-血管緊張系統的激活與參與。已報道在高氧致新生大鼠CLD肺組織勻漿中AngⅡ、血管緊張素轉化酶水平明顯上升[6];并且AT1R在高氧致CLD肺組織表達亦顯著增加[14]。本研究試圖探討AT1R拮抗劑losartan是否能阻止CLD的發生及其對高氧致CLD新生大鼠肺組織膠原沉積的影響。本研究的病理結果顯示,在高氧后期肺泡間隔明顯增寬、膠原沉積增加,losartan治療后肺泡間隔變薄、膠原纖維排列比較疏松;但肺泡直徑沒有明顯縮小,肺泡仍處于擴張狀態,次級隔同組Ⅰ相比仍明顯減少,呼吸膜面積并沒有明顯增加。進一步定量研究發現特異性AT1R拮抗劑losartan能夠抑制高氧致CLD新生大鼠肺組織ColⅠ蛋白和mRNA的表達,由此可以推測losartan雖可以抑制高氧CLD新生大鼠肺組織膠原的沉積,但是并不能逆轉高氧誘導的新生大鼠肺發育阻滯。因此,losartan并不能阻止高氧誘導CLD的發生。
[1]Marshall RP,Gohlke P,Chambers RC,et al.Angiotensin II and the fibroproliferative response to acute lung injury[J].Am J Physiol Lung Cell Mol Physiol,2004,286(1):L156-L164.
[2]Okada M,Suzuki K,Matsumoto M,et al.Effects of angiotensin on the expression of fibrosis-associated cytokines,growth factors,and matrix proteinsinhumanlungfibroblasts[J].JClinPharmTher,2009,34(3):288-299.
[3]Li X,Rayford H,Uhal BD.Essential roles for angiotensin receptor AT1a in bleomycin-induced apoptosis and lung fibrosis in mice[J].Am J Pathol,2003,163(6):2523-2530.
[4]Yao HW,Zhu JP,Zhao MH,et al.Losartan attenuates bleomycin-i nduced pulmonary fibrosis in rats[J].Respiration,2006,73(2):236-242.
[5]Molina-Molina M,Serrano-Mollar A,Bulbena O,et al.Losartan attenuates bleomycin induced lung fibrosis by increasing prostaglandin E2 synthesis[J].Thorax,2006,61(7):604-610.
[6]李玖軍,李洪鵬,薛辛東.高氧致慢性肺疾病新生大鼠肺組織腎素–血管緊張素系統和TGF-β1的動態變化及卡托普利的干預機制[J].中國醫科大學學報,2009,38(2):83-86.
[7]李玖軍,陳寧,薛辛東.高氧致CLD新生大鼠肺組織腎素–血管緊張素系統的動態變化及其意義[J].中國現代醫學雜志,2006,16(16):2423-2425.
[8]陳寧,李玖軍,薛辛東.洛沙坦對高氧致慢性肺疾病新生大鼠肺纖維化的影響[J].中國當代兒科雜志,2007,9(6):591-594.
[9]Marshall RP,Mc Anulty RJ,Laurent GJ.Angiotensin Ⅱ is mitogenic for human lung fibroblasts via activation of the type 1 receptor[J].Am J Respir Crit Care Med,2000,161(6):1999-2004.
[10]Wenzel S,Taimor G,Piper HM,et al.Redox-sensitive intermediates mediate angiotensin Ⅱ-induced p38 MAP kinase activation,AP-1 binding activity,and TGF-beta expression in adult ventricular cardiomyocytes[J].J FASEB,2001,15(12):2291-2293.
[11]Mancini GB,Khalil N.Angiotensin Ⅱ type 1 receptor blocker inhibitspulmonaryinjury[J].ClinInvestMed,2005,28(3):118-126.
[12]Keogh KA,Standing J,Kane GC,et al.Angiotensin Ⅱ antagonism fails to ameliorate bleomycin-induced pulmonary fibrosis in mice[J].Eur Respir J,2005,25(4):708-714.
[13]Chen CM,Chou HC,Hsu HH,et al.Transforming growth factorbeta1 upregulation is independent of angiotensin in paraquat-induced lung fibrosis[J].Toxicology,2005,216(2-3):181-187.
[14]陳寧,劉雪雁,富建華,等.高氧致新生大鼠慢性肺損傷肺組織血管緊張素Ⅱ1型受體表達的變化[J].中國醫科大學學報,2008,37(2):162-164.
猜你喜歡
中老年保健(2022年2期)2022-11-25 23:46:31
昆明醫科大學學報(2022年4期)2022-05-23 13:04:50
昆明醫科大學學報(2021年4期)2021-07-23 01:21:34
國際呼吸雜志(2019年21期)2019-11-25 09:52:20
國際呼吸雜志(2019年20期)2019-11-23 08:46:14
國際呼吸雜志(2019年8期)2019-04-29 09:15:22
國際呼吸雜志(2019年3期)2019-03-01 05:39:12
中國繼續醫學教育(2015年3期)2016-01-06 01:36:31
吉林大學學報(醫學版)(2015年3期)2015-12-17 07:47:38
天津醫科大學學報(2015年3期)2015-06-05 12:21:49
