黑果枸杞葉綠體基因組特征分析
2024-11-02 00:00:00柳迪杜雨李效雄馬瑞劉筠馬彥軍
西北農業學報 2024年10期


摘 要 為揭示黑果枸杞葉綠體基因組(cpDNA)特征及系統發育規律,采用二代測序技術對黑果枸杞葉綠體進行測序,并分析其基因組特征。結果表明:黑果枸杞cpDNA全長154 979 bp,其中包括1個大單拷貝區LSC,長度為85 984 bp,1個小單拷貝區SSC,長18 205 bp和2個反向互補重復區(IRs,25 395 bp)。共注釋到125個基因,其中,81個蛋白編碼基因(CDS),36個tRNA基因,8個rRNA基因。共檢測到60個核苷酸重復序列,有5種核苷酸類型,其中單核苷酸重復最多,為38個,占總體數的63.33%,基因序列為A/T。密碼子偏好性研究顯示,黑果枸杞cpDNA偏好G或C堿基,其中編碼亮氨酸(Leu)的密碼子最多,為2 154個,占總數的10.57%;編碼半胱氨酸(Cys)的密碼子最少,為219個。多態性分析結果顯示,IR區域表現出最高的保守性,而高變區在SSC區;SSR位點在葉綠體基因組中分布不均,多態性較高。系統發育結果表明,寧夏枸杞和中國枸杞聚為一類,與黑果枸杞、非洲枸杞匯聚在一起形成姐妹分支,四者之間的遺傳關系較近。
關鍵詞 黑果枸杞;葉綠體基因組;SSR;密碼子;系統發育分析
黑果枸杞(Lycium ruthenicum Murr)為茄科(Solanaceae)枸杞屬(Lycium),是一種多年生灌木,主要分布在中國西北荒漠地區,在中亞和歐洲等地也有分布,具有極高的藥用和食用價值[1-2]。黑果枸杞花色苷含量的測定原理主要通過其光學特性,目前常用的測定方法包括:色階法、消光系數法和pH示差法等[3]。阿力同·其米克等[4]通過ISSR分子標記法及DNA分析法研究黑果枸杞的遺傳多樣性,從60個引物中篩選出7個有效引物,結果顯示,黑果枸杞在物種水平的遺傳多樣性較高。Yun等[5]通過物理和酶的方法從黑果枸杞中提取多糖,發現表面枸杞多糖的結構與分離純化方法及其生長環境有關。Sina等[6]使用ImageJ程序測定不同基因型的枸杞葉片特征,研究表明,在同一物種的不同基因型之間,枸杞葉片的大小和形狀存在明顯差異。
隨著分子測序技術的迅速發展,植物系統發育研究從比較生理學和形態學開始向分子研究轉變[7]。葉綠體基因組具有相對分子量小、結構簡單且高度保守的特點,因此廣泛應用于分子系統發育研究[8]。安明珠等[9]在基于葉綠體基因,篩選鴨茅(Dactylis glomerata L .)葉綠體引物的研究中,從搜集的35對葉綠體引物中成功篩選出7對作為目的引物,為鴨茅的系統發育分析提供了依據。Yin等[10]通過采用Illumina高通量測序技術對野茄子(Solanum coagulans Forsk .)葉綠體基因組測序研究顯示,整個葉綠體基因組共注釋了128個編碼基因,包括83個蛋白編碼基因、37個轉移RNA基因和17個核糖體RNA基因;完整葉綠體基因組的長度為155 771 bp,其GC含量為37.76%。毛桂蓮等[11]通過鈣離子和過氧化氫對NaCl脅迫下枸杞葉片葉綠體功能的影響研究顯示,對氯化鈉脅迫的枸杞造成的傷害,外源Ca2+和H2O2具有一定緩解作用。Cui 等[12]對寧夏枸杞(L.barbarum)、中國枸杞 (L.chinense Mill)的葉綠體全基因組測序和分析,兩種枸杞葉綠體均注釋到133個基因,基因組全長分別為155 656 bp、155 745 bp,揭示了寧夏枸杞與中國枸杞的親緣關系。目前,有關黑果枸杞葉綠體的基因組研究較少,本研究以黑果枸杞葉片為材料,采用二代測序技術獲得黑果枸杞葉綠體全基因組序列,利用生物信息學相關軟件,分析其葉綠體基因組構成和特征,不僅豐富了黑果枸杞的遺傳信息,也為后續黑果枸杞的遺傳多樣性、種群歷史動態和枸杞親緣關系等發展研究奠定了基礎。
1 材料與方法
1.1 試驗材料
2017年采集民勤(地理坐標為39°01′N, 103°36′E,海拔1 309 m)1 a生野生黑果枸杞枝條,帶回學校,進行扦插育苗,2018年4月將扦插苗移栽到甘肅農業大學校內實習基地(地理坐標:38°28′N,106°16′E)。2022年7月,從長勢基本一致的不同黑果枸杞植株上隨機摘取成熟健康的葉片,沖洗干凈后,用液氮速凍處理,然后放于 -80 ℃超低溫冰箱中保存,備用。
1.2 葉綠體DNA提取和檢測
依據DNA提取試劑盒[生工生物工程(上海)股份有限公司,型號:B518731-0050]的提取步驟,將備用葉片放入研缽中充分研磨后提取黑果枸杞的cpDNA。經過除雜后,將檢測合格的 cpDNA進行純化、末端修復等過程,然后開始建立文庫,并且通過Illumina高通量測序獲得高質量數據(Clean Data)。
1.3 葉綠體全基因組序列拼接、組裝與注釋
使用SPAdes、GapFiller和PrInSeS-G軟件對基因組拼接、檢查與組裝,從而獲得完整葉綠體基因組。基于反向blast、參考基因組或內建數據庫進行比對,優化并提取拼接結果,注釋其CDS、tRNA、rRNA等基因元件。ARAGORN v1.2.38的tRNA注釋與blast結果進行矯正,最終匯總整理為完整的注釋結果。注釋完成后,提交到NCBI數據庫(https://www.ncbi.nlm.nih.gov/genbank/),利用OGDRAW軟件繪制黑果枸杞葉綠體基因組完整圖譜。
1.4 葉綠體基因組比較和系統發育分析
利用MISA軟件(https://webblast.ipk-gatersleben.de/misa/index.php)對枸杞的微衛星序列(SSR)進行分析和檢測。根據分布模式將黑果枸杞葉綠體基因組重復序列分為散在重復序列和串聯重復序列,使用在線軟件REPuter(https://bibiserv.cebitec.uni-bielefeld.de/reputer)分析枸杞葉綠體基因組的重復序列。利用CodonW1.4.2軟件對黑果枸杞葉綠體基因組密碼子偏好性(RSCU,relative synonymous codon usage)進行分析和統計。利用DnaSP軟件對黑果枸杞的葉綠體核苷酸多態性指數(nucleotide diversity,Pi)進行計算與分析。利用IRscope軟件(https://irscope.shinyapps.io/irapp/)繪制黑果枸杞、紅果枸杞、黃果枸杞、白果枸杞、煙草和擬南芥的葉綠體基因組邊界對比圖,比較枸杞種間與其他物種間的基因大小和分布差異。為探究黑果枸杞的系統進化關系,利用muscle軟件對22個葉綠體全基因組序列進行多重序列比對,使用raxml軟件和最大似然法(ML)構建系統發育樹。
2 結果與分析
2.1 葉綠體基因組結構與特征
將已測得的黑果枸杞葉綠體基因序列上傳到NCBI,從中獲取黑果枸杞葉綠體的基因序列號為OP846043。由表1可知,黑果枸杞葉綠體基因組的總長度為154 979 bp,GC含量為37.9%。黑果枸杞葉綠體基因組結構與大多數被子植物相同,呈四段式結構,由IRA、IRB、LSC和SSC組成,且4個區的長度存在差異,LSC區最長,SSC區最短;其中,LSC結構區長度為85 984 bp,SSC結構區長度為18 205 bp,2個反向互補重復區IR長度相等,總長為50 790 bp。
由圖1可知,黑果枸杞葉綠體基因組中共檢測到125個基因,其中,注釋有81個蛋白編碼基因(protein-coding genes,CDS),36個tRNA基因,8個rRNA基因。其中有17個基因在IR區重復,包含6個蛋白質編碼基因( rpl2、 rpl23、ndhB、 rps12、 rps7、 ycf2),4個rRNA基因( rrn16、 rrn23、 rrn4.5、 rrn5),7個tRNA基因(trnA-UGC、trnI-CAU、trnI-GAU、trnL-CAA、trnN-GUU、trnR-ACG、trnV-GAC);SSC區包含11個蛋白質編碼基因,1個tRNA基因;LSC區包含62個蛋白質編碼基因,21個tRNA基因。
如表2所示,葉綠體基因主要包括光合作用和自我翻譯功能,依據基因功能可劃分為光合系統基因,遺傳系統基因,其他基因和未知功能基因。其中ndhA、petB、atpF、trnG-UCC等12個基因含有1個內含子,clpP和 ycf3則含有2個內含子,而內含子比外顯子更容易在進化中喪失功能。與核糖體RNA的合成有關的4個基因都位于IR區,且存在2個拷貝。此外,有12個基因存在2個拷貝,分別為: rpl2, rpl23,trnI-CAU,trnI-GAU,trnA-UGC,trnR-ACG,trnL-CAA,trnV-GAC, ycf2、ndhB、trnN-GUU、 rps7。
2.2 葉綠體基因組重復序列分析
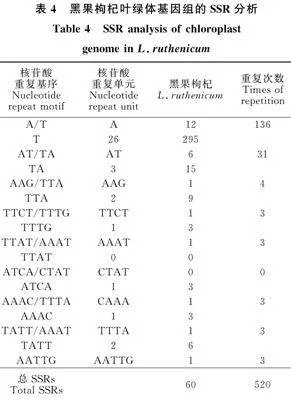
黑果枸杞葉綠體基因組的簡單重復序列(SSR)分析結果如表3和表4所示:黑果枸杞的核苷酸重復序列有60個,在5種核苷酸類型中,單核苷酸重復最多,為38個,占總體數的 63.33%,基因序列為A/T;其次是二核苷酸和四核苷酸序列,均為9個,各占總數的15%,其中,二核苷酸重復包含AT/TA基因序列,四核苷酸重復包含TTCT/TTTG、AAAT/TATT、TTAT/TTTA、CAAA/AAAC和CTAT/ATCA 4種基因序列類型;三核苷酸重復數為3個,占總體數的5%,重復包含AAG/TTA基因序列;五核苷酸重復數是1個,占總體數的1.70%,包含AATTG基因序列。
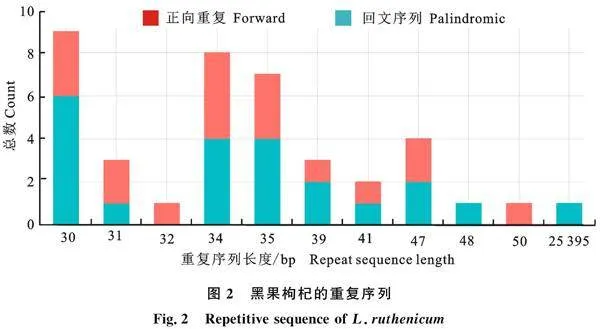
散在重復序列主要包括4種類型:正向重復(forward,F)、回文重復(palindrome,P)、反向重復(reverse,R)和互補重復(complement,C)。如圖2所示,黑果枸杞葉綠體的散在重復序列包含回文序列(palindromic)和正向重復序列(forward)2種,共40條,其中有22條回文序列和18條正向重復序列;對黑果枸杞的重復序列分析顯示大多數重復序列分布在基因間隔區或內含子區域,且絕大多數序列的長度為30~50 bp。
2.3 葉綠體基因組密碼子偏好性分析
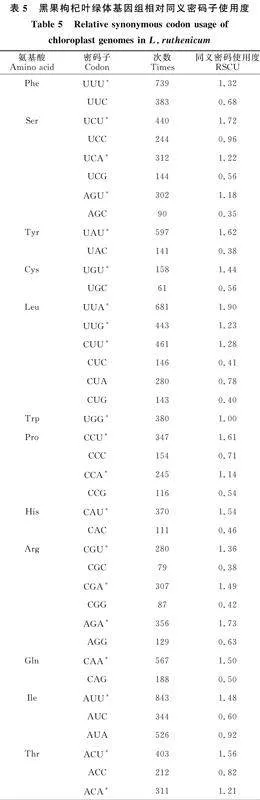
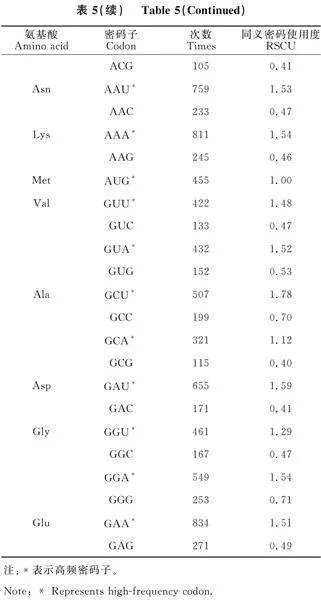
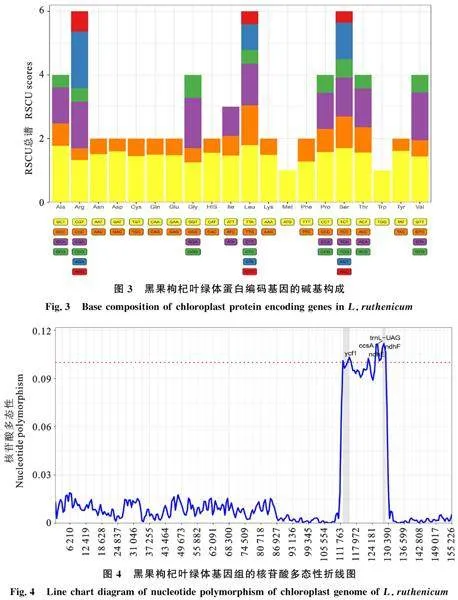
對黑果枸杞葉綠體密碼子分析結果如表5所示,檢測到所有密碼子的出現次數為20 370(終止子除外),其中編碼亮氨酸(Leu)的密碼子為UUA、UUG、CUU、CUC、CUA、CUG,出現次數為2 154,占總數的10.57%;編碼半胱氨酸(Cys)的密碼子為UGU、UGC,出現次數最少,為219,占總數的1.08%。相對同義密碼子相對使用度的值大于1時,說明密碼子在蛋白質編碼的使用上具有偏好性。使用度最高的是UUA,為 1.90,使用度最低的是AGC,為0.35;有31個密碼子的RSCU值大于1,其中,有28個密碼子的堿基構成以A或U結尾,3個密碼子的堿基構成以G或C結尾。
葉綠體蛋白質編碼基因的RSCU值的大小是衡量密碼子偏好性的標準。如圖3所示,黑果枸杞葉綠體蛋白編碼基因主要偏好以A、T結尾的密碼子,且T的使用率大于A。以亮氨酸(Leu)和絲氨酸(Ser)為例,編碼Leu的密碼子共有6個(TTA、TTG、CTT、CTC、CTA、CTG),其中TTA的RSCU值最大,為1.9,CTC和CTG的RSCU值最小,分別為0.41和0.40,說明黑果枸杞葉綠體在編碼Leu時偏好使用密碼子TTA,而盡量避開CTC和CTG兩個密碼子;編碼Ser的密碼子也有6個(TCT、TCC、TCA、TCG、AGT、AGC),但測序結果顯示主要使用TCT、TCA、AGT(RSCU值均大于1),尤其是TCT的使用率最高,而AGC的使用率最低,為0.35。
2.4 葉綠體基因組的核苷酸序列多態性分析
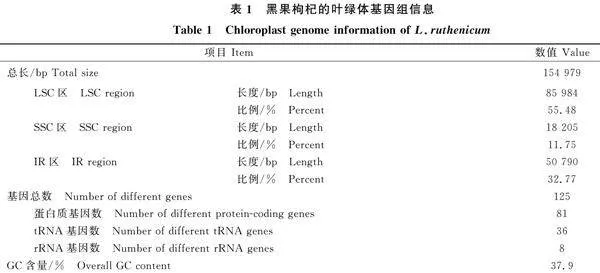
對黑果枸杞葉綠體基因組序列進行多態性分析,結果如圖4所示,用核苷酸變異性(Pi)表示,在黑果枸杞葉綠體基因組的4個典型區域中,IR區域表現出最高的保守性,而高變區在SSC區;黑果枸杞葉綠體CDS中突變率最高的5個基因分別為 ycf1、ndhE、ccsA、trnL-UAG和ndhF,Pi值依次為0.103 31、0.102 55、0.111 31、0.111 49和0.111 75,這些區域可能包含關于葉綠體基因組中進化速率較快的位點信息。
2.5 葉綠體基因組IR區邊界分析
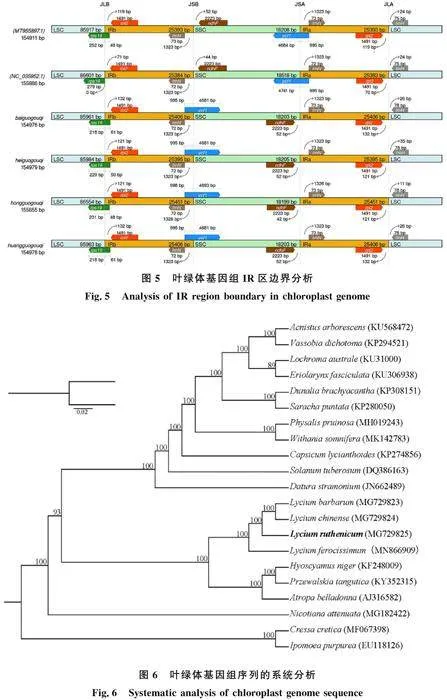
為探究葉綠體基因組的IR區與LSC、SSC區之間連接區域的詳細信息,對模式植物擬南芥(MT955897.1)、煙草(NC_035952.1)和4種枸杞屬植物的葉綠體進組進行分析。結果如圖5所示,6個葉綠體基因組中IR區域的長度范圍為 25 384~25 451 bp,顯示出適度的擴張。在這6個物種的葉綠體基因組中,除煙草的LSC區明顯擴張外,其他物種的IRb/LSC的連接區域均處于 rps19基因的編碼區,導致了3′端基因的重復,而這種重復在IRb/LSC的連接區域產生了長度不等(48~61 bp)的 rps19假基因;此外,4種枸杞屬植物的IRb/SSC邊界和2種模式植物的IRa/SSC邊界均存在于 ycf1基因的編碼區,煙草和擬南芥在IRb/SSC邊界產生的 ycf1假基因長度均為995 bp,4種枸杞屬植物在IRa/SSC邊界產生的 ycf1假基因長度不等(995~998 bp)。在這些葉綠體基因組中, rpl2基因長度為1 491 bp,分別位于IRb區域靠近IRb/LSC邊界71~132 bp和IRa區域靠近IRa/LSC邊界 70~132 bp的位置;trnN基因分別位于IRb區域靠近IRb/SSC邊界和IRa區域靠近IRa/SSC邊界72 bp(擬南芥為73 bp)的位置;trnH基因僅存在于LSC區,距離IRa/LSC邊界11~26 bp;ndhF基因只存在于SSC區域,擬南芥和煙草靠近IRb/SSC邊界的距離分別為52 bp、44 bp,其他4種枸杞屬植物則距離IRa/SSC邊界52 bp(紅果枸杞為42 bp)。[JP]
2.6 葉綠體基因組的系統進化分析
為了確定黑果枸杞的系統進化發育關系,本研究將黑果枸杞與茄科以及2個外類物種等共21個cpDNA序列構建ML系統發育樹。發育樹如圖6所示,21個物種可分為2類,第1類是外類群組,堿草(Cressa cretica)和圓葉牽牛(Ipomoea purpurea)聚為1類,第2類由19個茄科物種組成;第2類群又可分為2個小類群,其中煙草(Nicotiana attenuata)自為1類,其余的茄科植物為一類。寧夏枸杞(Lycium barbarum)與中國枸杞(Lycium chinense)聚為一類,與黑果枸杞 (Lycium ruthenicum)、非洲枸杞(Lycium ferocissimum)匯聚在一起形成姐妹分支,支持率較高。枸杞屬植物與天仙子(Hyoscyamus niger)、馬尿脬(Przewalskia tangutica)和顛茄(Atropa belladonna)匯聚成的姐妹分支聚為一個大分支,說明枸杞屬植物與這3個屬的植物遺傳關系 較近。
3 討 論
目前,因基因組測序技術的廣泛運用和葉綠體基因組的深入研究,使其數據庫迅速擴大,從而為探究分子進化的過程提供了基礎,與枸杞核基因組相比,葉綠體基因組更具有顯著的優勢,因此,本試驗對黑果枸杞葉綠體的全基因組序列進行研究。試驗測得黑果枸杞的cpDNA全長為154 979 bp,LSC長度為85 984 bp,SSC為 18 205 bp,IR為25 395 bp,與已報道的高等植物的葉綠體基因組數據相符[13]。有關枸杞屬葉綠體基因組測序研究的報道較少,目前只有2種枸杞屬植物被報道,分別為中國枸杞[14] 、寧夏枸杞 [15]。比較這3種枸杞葉綠體基因組的組成和特征發現,寧夏枸杞和中國枸杞葉綠體基因組總GC含量為37.8%;黑果枸杞葉綠體基因組GC含量為37.9%,而GC的含量可反應葉綠體基因組組成特征,在同屬內枸杞的葉綠體基因組的進化速度慢,表現出的保守性較高。不同植物葉綠體的tRNA基因數目存在較大差異,如砂仁[16](Amomum villosum Lour.)葉綠體基因組中注釋113個基因,包括79個蛋白編碼基因,30個tRNA個基因和4個rRNA基因;山楂[16](Crataegus pinnatifida Bunge.)注釋到112個基因,包括78個蛋白編碼基因、30個tRNA基因和4個rRNA基因。對測得的黑果枸杞葉綠體基因組進行基本功能注釋,共注釋到125個基因,檢測到81個蛋白編碼基因,36個tRNA基因,8個rRNA基因,而出現這種差異的原因是不同植物之間葉綠體基因本就存在較大差異。黑果枸杞的編碼基因主要分布在LSC區,大多數基因只含有1個內含子,這與其他植物研究結果一致[17]。SSR序列常見于動物和植物中,由于DNA在進行復制時 ,堿基容易發生錯位,因此使得SSR序列具有豐富的多態性,為生物的進化和系統進化提供依據[18]。枸杞的散在重復序列主要有回文序列(Palindromic)和正向重復序列(Forward)2種類型[14],在39~48之間,并且回文序列的重復次數較多,黑果枸杞葉綠體基因組含22條回文序列和18條正向重復序列。在黑果枸杞的葉綠體基因組中,檢測出60個SSR序列,共有5種類型(除了這5種重復序列外,其他所有重復序列都位于相同的基因或基因間區域),其中大多數為單核苷酸重復序列,沒有檢測到六核苷酸序列,且主要由多聚腺苷酸(polyA)和多聚胸腺嘧啶(polyT)重復序列組成[19]。本研究中的重復序列差異,有助于枸杞植物的探索和利用。黑果枸杞SSR位點在葉綠體基因組中分布不均,多態性較高,為人們研究枸杞葉綠體基因組片段的進化,以及SSR分子標記技術的開發提供理論依據。
密碼子的偏好性是由基因突變和物種進化等多種因素作用下形成的[20]。在植物基因中,不論在同一物種還是不同物種之間,不同密碼子的出現頻率有明顯差異。RSCU值是衡量密碼子偏好性的一個重要參數,RSCU值越大,則表示該密碼子在基因表達中的使用頻率越高,反之則越低;當RSCU 值等于1時,認為該密碼子的使用情況僅受基因突變的影響[21]。本研究中黑果枸杞葉綠體基因組有31個密碼子的RSCU值大于1,說明這31個密碼子在蛋白質基因編碼上具有偏好性,其中以A或U結尾的密碼子占90.32%,這與番茄等植物的研究結果一致[22];而在山楂屬[23]的葉綠體基因組中,最優密碼子的數目為17~20個,兩者之間的差異可能與植物種類以及選入的基因數量有關。比較密碼子使用頻率之間的差異,在黑果枸杞中亮氨酸(Leu)、精氨酸(Arg)、絲氨酸(Ser)RSCU同義密碼子使用度最大,由此推測,黑果枸杞中這3種氨基酸的含量可能較高。普遍認為IR區是葉綠體基因組中最保守的部位,而形成植物葉綠體基因組長度變化的主要原因是IR區邊緣的收縮和擴張[24]。研究表明,葉綠體基因組的演化和系統發育分析的過程可由 ycf1、ndhF基因的特征分析[25]。本試驗研究發現,不同物種葉綠體基因組的IR區與LSC、SSC區之間連接區域的編碼基因存在相似的規律,但仍存在一定差異,例如 ycf1和ndhF基因在不同的連接區域編碼。在枸杞屬植物的葉綠體基因組IR區分析中發現,屬內植物的連接區域及基因表達具有高度相似性,這與丁香屬[26]植物的研究結果也類似。核苷酸多態性可以用來評價群體基因組遺傳多樣性水平,本研究得出在黑果枸杞葉綠體基因組中,IR區域表現出最高的保守性,而高變區在SSC區,其中ndhF基因的多態性最高,且 ycf1、ccsA和ndhE等基因的突變率也比較高,這些突變位點可為枸杞屬內物種鑒定和系統進化分析提供更有價值的信息,這與南洋杉科葉綠體基因組在基因組特征的各個方面均高度保守的特征相同[27]。系統發育基本組學研究是通過分子數據來探討生物之間的發育規律的;系統發育對于研究進化歷史對生態群落中物種組裝的影響以及物種組合系統發育結構的地理和生態模式至關重要[28-29]。由于cpDNA具有序列保守、結構穩定等特點[30],因此,基于cpDNA進行的系統發育研究得到了很好的發展。本研究以測得黑果枸杞葉綠體基因組全部序列聯合NCBI下載的茄科植物以及外類群組植物的葉綠體基因組序列構建了進化樹,結果表明,該進化樹的分辨率較高,各節點也獲得了較高的支持率,茄科內屬間呈現出較為明確的發育關系,枸杞屬內呈明顯的姐妹關系,與Wang等 [31]研究的枸杞(寧夏枸杞)的進化關系一致。
4 結 論
綜上所述,黑果枸杞cpDNA全長154 979 bp,共注釋到125個基因,其中,包含有81個蛋白編碼基因,36個tRNA基因,8個rRNA基因,黑果枸杞cpDNA偏好G或C堿基。系統發育結果表明,黑果枸杞與非洲枸杞、寧夏枸杞和中國枸杞等枸杞屬植物親緣關系較近。本研究豐富了黑果枸杞的研究內容,為進一步研究黑果枸杞系統進化和遺傳育種提供了理論依據。
參考文獻 Reference:
[1] 萬國海.枸杞的生物學特性、栽培管理及采收加工[J].種植與環境,2013(4):214.
WAN G H.Biological characteristics,cultivation management and harvesting and processing of Lycium barbarum L.[J].Modern Animal Husbandry Science & Technology,2013(4):214.
[2]YU ZH L,XIA M Q,AN J P,et al.A comprehensive review on the ethnobotany,phytochemistry,pharmacology and quality control of the genus Lycium in China[J].Food & Function,2023,14:2998-3025.
[3]閆亞美,冉林武,曹有龍,等.黑果枸杞花色苷含量測定方法研究[J].食品工業,2012,33((6):145-147.
YAN Y M,RAN L W,CAO Y L,et al.Determine the total anthocyanins in Lycium ruthenicum Murr.by different methods[J].The Food Industry,2012,33((6):145-147.
[4]阿力同·其米克,王青鋒,楊春鋒,等.新疆產藥用植物黑果枸杞遺傳多樣性的ISSR分析[J].植物科學學報,2013, 31(5):517-524.
ALITONG QIMIKE,WANG Q F,YANG CH F,et al.ISSA analysis on genetic diversity of medically important Lycium ruthenicum Murr.in Xinjiang.[J].Plant Science Journal, 2013,31(5):517-524.
[5]YUN D W,YAN Y M,LIU J.Isolation,structure and biological activity of polysaccharides from the fruits of Lycium ruthenicum Murr:A review[J].Carbohydrate Polymers,2022,291:119618.
[6]SINA C,FLAVIA S,MANUELA M.Determination of leaf characteristics in different medlar genotypes usingthe image program[J].Horticultural Science,2020,47(2):117-121.
[7]SOMSSICH MARC.The dawn of plant molecular biology:how three key methodologies paved the way[J].Current Protocols,2022,2(4):e417.
[8]潘 登,高宏波.葉綠體DNA的發現歷程[J].生物學通報,2012,47(7):53-55.
PAN D,GAO H B.Discovery process of chloroplast DNA[J].Bulletin of Biology,2012,47(7):53-55.
[9]安明珠,趙世偉,朱文露,等.鴨茅葉綠體引物篩選[J].草學,2022(1):30-36,47.
AN M ZH,ZHAO SH W,ZHU W L,et al.Screening of Dactylis chloroplast primers[J].Journal of Grassland and Forage Science,2022(1):30-36,47.
[10] YIN M Y,YU Y A,GONG Y J,et al.The complete chloroplast genome of; Solanum sisymbriifolium; (Solanaceae),the wild eggplant[J].Mitochondrial DNA Part B,2022,7(5):886-888.
[11]毛桂蓮,付曉輝.外源Ca2+和H2O2對NaCl脅迫下枸杞離體葉片葉綠體中活性氧代謝的影響研究[J].干旱地區農業研究,2009,27(5):191-195.
MAO G L,FU X H.Effects of exogenous Ca2+ and H2O2 on active oxygen metabolism of isolated leaves of Lycium barbarum L.chloroplast under NaCl stress[J].Agricultural Research in the Arid Areas,2009,27(5):191-195.
[12]CUI Y,ZHOU J,CHEN X,et al.Complete chloroplast genome and comparative analysis of three Lycium (Solanaceae) species with medicinal and edible properties[J].Gene Reports,2019,10:0464.
[13]ZHANG T W,FANG Y J,WANG X M,et al.The complete chloroplast and mitochondrial genome sequences of Boea hygrometrica:insights into the evolution of plant organellar genomes[J].PloS One,2012,7(1):e30531.
[14]YANG Z,HUANG Y,AN W,et al.Sequencing and structural analysis of the complete chloroplast genome of the medicinal plant Lycium chinense Mill[J].Plants,2019, 8(4):87.
[15]劉晶星.寧夏枸杞和黑果枸杞葉綠體全基因組測序及基因注釋分析[D].北京:中國科學院北京基因組研究所,2013.
LIU J X.De novo assembly and gene annotation of the chloroplast genomes of Lycium barbarum and Lycium ruthenicum[D].Beijing:Beijing Institute of genome research,Chinese Academy of Sciences,2013.
[16]崔英賢.藥食兩用藥材砂仁、枸杞、山楂和姜基原植物葉綠體基因組結構解析[D].北京:北京協和醫學院,2020.
CUI Y X.Structural analysis of complete chloroplast genome of medicinal and edible original plants of Amomum,Chinese Wolfberry,Hawthorn and Ginger[D].Beijing:Peking Union Medical College,2020.
[17]NI Z X,YE Y J,BAI T D,et al.Complete chloroplast genome of Pinus massoniana (Pinaceae):Gene rearrangements,loss of ndh genes,and short inverted repeats contraction,expansion[J].Molecules,2017,22(9):1528.
[18]JANSEN R K,SASKI C,LEE S,et al.Complete plastid genome seque nces of three Rosids (Castanea,Prunus,Theobroma):evidence for at least two independent transfers of rpl22 to the nucleus[J].Molecular Biology and Evolution,2011,28(1):835-847.
[19]ZHANG Y J,DU L W.The complete chloroplast genome sequences of five Epimedium species:lights into phylogenetic and taxonomic analyses[J].Frontiers in Plant Science,2016,7(696):1-11.
[20]HLNE CHIAPELLO A B,FRDRIQUE LISACEK B,et al.Codon usage and gene function are related in sequences of Arabidopsis thaliana[J].Gene,1998,209(1/2):1-38.
[21]SONG H,LIU J,CHEN T,et al.Synonymous codon usage pattern in model legume Medicago truncatula[J].Journal of Integrative Agriculture,2018,17(9):2074-2081.
[22]LI N,LI Y Y,ZHENG C C,et al.Genome-wide comparative analysis of the codon usage patterns in plants[J].Genes&Genomics,2016,38(8):723-731.
[23]趙振寧,孫浩田,宋雨茹,等.山楂屬植物葉綠體基因組特征與密碼子偏好性分析[J].江蘇農業學報,2023,39(2):504-517.
ZHAO ZH N,SUN H T,SONG Y R,et al.Chloroplast genome characteristics and codon usage bias analysis of Crataegus L.[J].Jiangsu Journal of Agricultural Sciences,2023,39(2):504-517.
[24]WANG Q,ZHANG CH H,WANG CH J,et al.Complete chloroplast genome data for Mimosa diplotricha and Mimosa diplotricha var.inermis from China[J].Data in Brief,2023,48:109045.
[25]JOEY S,EDGAR B L,EDWARD E S,et al.Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms:the tortoise and the hare III[J].American Journal of Botany,2007,94(3):275-288.
[26]張 航.丁香屬3種植物完整葉綠體基因組與系統發育研究[D].哈爾濱:東北林業大學,2022,001431.
ZHANG H.Complete chloroplast genomes and phylogeny of three plants of the Syringa[D].Harbin:Northeast Forestry University,2022,001431.
[27]VAN BINH NGUYEN,VO NGOC LINH GIANG,NOMAR ESPINOSA WAMINAL,et al.Comprehensive comparative analysis of chloroplast genomes from seven Panax species and development of an authentication system based on species-unique single nucleotide polymorphism markers[J].Journal of Ginseng Research,2020,44(1):135-144.
[28]TANGJ Y,WEI R,ZHANG X CH,et al.Mitogenome-based phylogenomics provides insights into the positions of the enigmatic sinensis group and the sanguinolenta group in Selaginellaceae (Lycophyte)[J].Molecular Phylogenetics and Evolution,2023,179:107673.
[29]YU M,ZHANG D,ZHAO X M.Sequencing and phylogenomics of the complete mitochondrial genome of Allodiplogaster sp.(Rhabditida:Diplogasteridae):a new gene order and its phylogenetic implications[J].Gene,2022,840:146761.
[30]CASTROA A,NUNES R,CARVALHO L R,et al.Chloroplast genome characterization of Uncaria guianensis and Uncaria tomentosa and evolutive dynamics of the Cinchonoideae subfamily[J].Scientific Reports,2023,13(1):8390.
[31]WANG C P,JIA G L,ZHU L ZH,et al.The complete chloroplast genome sequence of Lycium ruthenicum(Solanaceae),a traditional medicinal plant in China[J].Mitochondrial DNA Part B,2019,4(2):2495-2496.
Analysis of Chloroplast Genomic Characteristics of L.ruthenicum Murr
LIU Di1,DU Yu1,LI Xiaoxiong 2,MA Rui1,LIU Yun1 and MA Yanjun1
(1.Forestry College,Gansu Agriculture University,Lanzhou 730070,China; 2.LanzhouResources & Environment Voc-Tech University,Lanzhou 730030,China )
Abstract Tounveil the chloroplast genome (cpDNA) characteristics and phylogenetic development of Lycium ruthenicum,we sequenced the chloroplast of Lycium ruthenicum using NGS technology,and analyzed its genomic characteristics.The results showed that the total length of the cpDNA of Lycium ruthenicum was 154 979 bp,comprising a large single copy region LSC with a length of 85 984 bp,a small single copy region SSC with a length of 18 205 bp,and two reverse complementary repeat regions (IRs) totaling 25 395 bp. Annotation identified a total of 125 genes,including 81 protein coding genes (CDS),36 tRNA genes,and 8 rRNA genes. We detected 60 nucleotide repeat sequences, with five nucleotide types showing the highest single nucleotide repeat of 38,accounting for 63.33% of the total number. The gene sequence was A/T. Codon preference analysis showed a preference for G or C bases,with 2 154 codons encoding Leucine (Leu) accounting for 10.57% of the total,while the least encoded codon was Cysteine (Cys) with 219 occurrences.Polymorphism analysis showed that the IR region exhibited the highest conservatism,while the SSC region was hypervariable; the SSR loci were unevenly distributed and exhibited high polymorphism in the chloroplast genome. The phylogenetic analysis indicated that Lycium barbarum and Lycium chinense clustered together forming sisters branches with Lycium barbarum and Lycium ferocissimum,indicating a close genetic relationship among the four species.
Key words Lycium ruthenicum,; Chloroplast genome; SSR; Codon; Phylogenetic analysis
Received 2023-08-16 Returned 2023-09-19
Foundation item Research Project of Lanzhou Resources & Environment Voc-Tech University (No.X2022ZD-05); University Industry Support Plan of Gansu Province (No.2023CYZC-46); Lanzhou Science and Technology Plan (No.2022-2-22).
First author LIU Di,male,master student.Research area:investigation,collection,preservation,and research of plant germplasm resources. E-mail:18193143002@163.com
Corresponding author MA Yanjun,male,Ph.D,professor.Research area:investigation,collection,preservation,and research of plant germplasm resources.E-mail:mayanjun@gsau.edu.cn
(責任編輯:潘學燕 Responsible editor:PAN Xueyan)
基金項目:蘭州資源環境職業技術大學重點研究項目(X2022ZD-05);甘肅省高校產業支撐計劃(2023CYZC-46);蘭州市科技計劃(2022-2-22)。
第一作者:柳 迪,男,碩士研究生,從事植物種質資源調查收集、保存與研究。E-mail:18193143002@163.com
通信作者:馬彥軍,男,博士,教授,主要從事植物種質資源調查收集、保存與研究。E-mail:mayanjun@gsau.edu.cn
