高效液相色譜法測定鹽酸羥考酮緩釋片中鹽酸羥考酮含量*
2024-02-25 04:07:20曾令高孫懷敬程浩男尹可欣林美齡熊莉平
中國藥業 2024年3期
馬 睿,曾令高,羅 娟,孫懷敬,程浩男,尹可欣,林美齡,熊莉平
(1.西南藥業股份有限公司,重慶 400038;2.重慶市食品藥品檢驗檢測研究院·國家藥品監督管理局麻醉精神藥品質量監測重點實驗室,重慶 401121)
腫瘤患者初期疼痛發生率約為25%,晚期疼痛發生率為60%~80%[1]。癌痛可能會引起或加重患者的焦慮、抑郁、乏力、失眠、食欲減退等癥狀,嚴重影響患者的日常活動、自理能力、交往能力及整體生活質量[1-2]。世界衛生組織(WHO)認定阿片類鎮痛藥為三階梯癌痛治療的常規用藥,能有效緩解中度至重度疼痛,是治療急性疼痛的一線藥物,但具有成癮性[3]。且使用即釋阿片類藥物服藥次數大大增加[4],患者擔心藥物成癮性,導致用藥依從性降低。鹽酸羥考酮是由生物堿蒂巴因植物衍生物制成的半合成阿片類藥物,自1917年開始應用于臨床,主要作用于中樞神經系統和平滑肌,對μ,κ,δ 受體具有激動劑活性[5-6],鎮痛作用強。鹽酸羥考酮緩釋片具有雙相釋放特點,38%首先快速釋放,其余62%隨后緩慢持續釋放[7-8],能在口服后1 h 內快速鎮痛,并持續12 h鎮痛,既起到了鎮痛效果,又大大降低了成癮性。故控制鹽酸羥考酮緩釋片中鹽酸羥考酮含量至關重要。為此,本研究中建立了測定鹽酸羥考酮緩釋片中鹽酸羥考酮含量的高效液相色譜法。現報道如下。
1 儀器與試藥
1.1 儀器
Agilent 1260 型高效液相色譜儀(美國Agilent 公司);Waters e2695 型高效液相色譜儀(美國Waters 公司);HT-800 型超聲波清洗機(海騰超聲電子設備有限公司,功率為800 W,頻率為25~40 kHz);ME204E型電子天平(精度為萬分之一),XPE26 型電子天平(精度為百萬分之一),均購自瑞士Mettler-Toledo公司。
1.2 試藥
鹽酸羥考酮對照品(批號為171271-201501,含量為94.9%),枸櫞酸三乙酯對照品(批號為190096-201802,含量為99.5%),均購自中國食品藥品檢定研究院;羥考酮雜質B對照品(消旋體,批號為2005604-3,含量為99.58%,含雜質B1及B22種異構體),羥考酮雜質C 對照品(批號為2005609-1,含量為99.19%),鹽酸羥考酮原料(批號為CP0012103005),均購自甘肅普瑞制藥有限公司;乳糖(德國美劑樂兩合公司);十八醇(江西阿爾法高科藥業有限公司);鹽酸羥考酮緩釋片(西南藥業股份有限公司,批號分別為Y211201,Y220901,Y221001,規格為每片10 mg);庚烷磺酸鈉(色譜純,山東禹城市中美色譜產品廠);磷酸(分析純,重慶川東化工<集團>有限公司);甲醇、乙腈(色譜純,賽默飛世爾科技<中國>有限公司);水為超純水。
2 方法與結果
2.1 色譜條件與系統適用性試驗
色譜柱:Agilent Eclipse XDB-C18柱(150 mm ×4.6 mm,5 μm);流動相:1.1 g/L 1-庚烷磺酸鈉溶液(pH 2.0)-乙腈-甲醇(750∶95∶155,V/V/V);流速:1.2 mL/min;檢測波長:230 nm;柱溫:40 ℃;進樣量:20 μL。系統適用性溶液在此色譜條件下,色譜峰的分離度大于1.5,理論板數按羥考酮峰計為4 886。
2.2 溶液制備
取鹽酸羥考酮、羥考酮雜質B、羥考酮雜質C 對照品各適量,精密稱定,加流動相溶解,稀釋成每1 mL 約含鹽酸羥考酮80μg、雜質B 和C 均為1.6μg 的系統適用性溶液。取枸櫞酸三乙酯、乳糖、十八醇各適量,精密稱定,加流動相溶解,稀釋成每1 mL 含乳糖、枸櫞酸三乙酯、十八醇均為1 mg 的空白輔料溶液。取鹽酸羥考酮對照品適量,精密稱定,加流動相溶解,稀釋成每1 mL約含鹽酸羥考酮80μg 的對照品溶液。取樣品10 片,除去包衣,精密稱定,研磨至極細粉末,精密稱取適量(約相當于鹽酸羥考酮8 mg),置100 mL 容量瓶中,加流動相適量,超聲使鹽酸羥考酮溶解完全,放冷,用流動相定容,搖勻,濾過,即得供試品溶液。
2.3 方法學考察
專屬性試驗:精密量取2.2項下空白輔料溶液及系統適用性溶液各適量,按2.1 項下色譜條件進樣測定,記錄色譜圖。系統適用性溶液色譜中,各成分分離度良好,且輔料無干擾。詳見圖1。
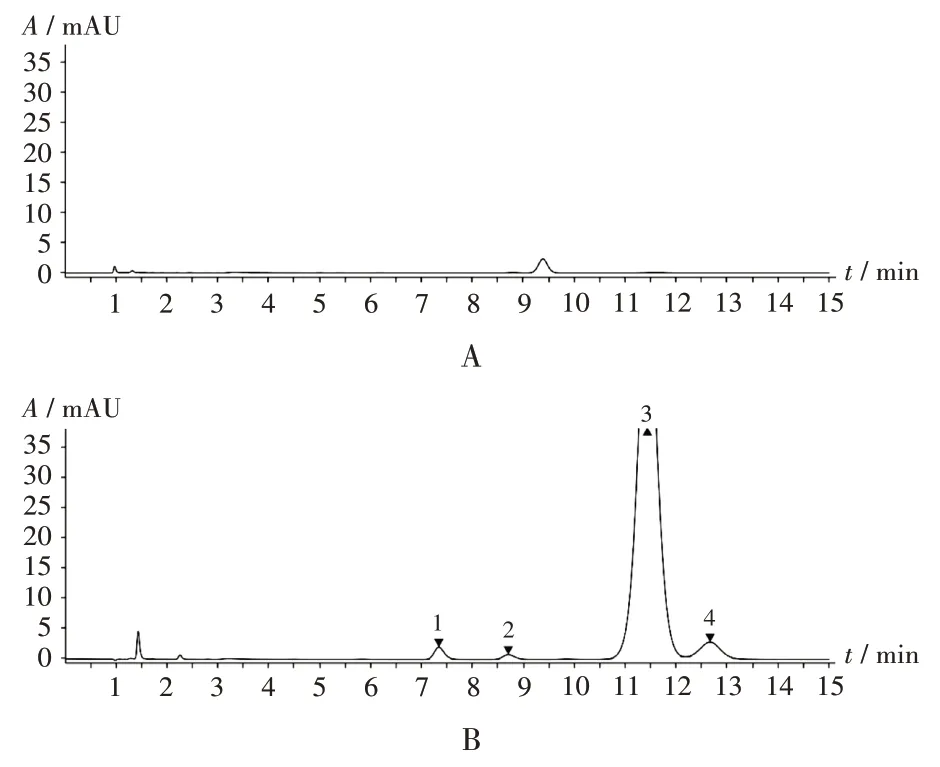
1.羥考酮雜質B1 2.羥考酮雜質B2 3.羥考酮 4.羥考酮雜質CA.空白輔料溶液 B.系統適用性溶液圖1 專屬性試驗高效液相色譜圖1.Oxycodone impurity B1 2.Oxycodone impurity B2 3.Oxycodone 4.Oxycodone impurity CA.Excipients-blank solution B.System suitability solutionFig.1 HPLC chromatograms of the specific test
定量限確定:取鹽酸羥考酮原料,按2.2 項下方法制備濃度為25%的線性溶液,按2.1項下色譜條件進樣測定,以信噪比(S/N)為10倍時的質量濃度為定量限。結果鹽酸羥考酮定量限為50.75 ng/mL。
線性關系考察:取鹽酸羥考酮原料,按2.2項下方法制備濃度分別為25%,100%,150%,200%,250%的系列線性溶液,精密量取各線性溶液及定量限溶液(質量濃度為50.75 ng/mL),按2.1項下色譜條件進樣測定,以羥考酮的質量濃度(X,μg/mL)為橫坐標、峰面積(Y)為縱坐標進行線性回歸,得回歸方程Y=20.884X+0.616 9(R2=1.000 0,n=6)。結果表明,羥考酮的質量濃度在0.047~189.920μg/mL范圍內與峰面積線性關系良好。
精密度試驗:精密量取2.2項下對照品溶液2份及供試品溶液1份,按2.1項下色譜條件分別進樣測定5次、2次、6次,記錄色譜圖。結果對照品溶液峰面積比值的RSD為0.52%,供試品溶液保留時間的RSD為0.01%(n=6),峰面積的RSD為0.11%(n=6),表明儀器精密度良好。
重復性試驗:取樣品(批號為Y211201)適量,按2.2項下方法制備供試品溶液6 份,由2 名操作人員在不同時間用不同儀器重復上述操作,計算鹽酸羥考酮的含量。結果2 名操作人員的RSD分別為0.33%(n=6)和0.78%(n=6),鹽酸羥考酮的平均含量為100.36%,RSD為0.62%(n=12),表明方法重復性良好。
穩定性試驗:取2.2 項下供試品溶液適量,于室溫放置0,6,24,48 h 時按2.1 項下色譜條件進樣測定,記錄峰面積,并計算含量。結果平均含量為100.16%,RSD為0.17%(n=4),表明供試品溶液在室溫放置48 h 內穩定性良好。
耐用性試驗:取樣品(批號為Y211201),按2.2 項下方法制備供試品溶液,分別考察不同的檢測波長(228,232 nm),柱溫(38,42 ℃),流速(1.1,1.3 mL/min),甲醇和乙腈體積分數(1±5%),緩沖液pH 值(1.8,2.2),色譜柱[Agilent Eclipse XDB-C18柱(150 mm ×4.6 mm,5 μm),Agilent Zorbax SB-C18柱(150 mm ×4.6 mm,5μm)],色譜儀(Agilent 1260 型、Waters e2695型)對樣品提取效果的影響。結果當測定條件發生微小變動時,均能達到分離要求,各條件下鹽酸羥考酮平均含量為100.09%,RSD為0.81%,能滿足測試要求。
回收試驗:取鹽酸羥考酮原料,照處方與工藝制備樣品,鹽酸羥考酮按處方量的80%,100%,120%投料,各平行3份,按2.2項下方法制備供試品溶液,按2.1項下色譜條件進樣測定,計算回收率。結果見表1。

表1 回收試驗結果(n=9)Tab.1 Results of the recovery test(n=9)
2.4 樣品含量測定
取3 批(批號分別為Y211201,Y220901,Y221001)樣品,按2.2 項下方法制備供試品溶液,按2.1 項下色譜條件進樣測定,每批樣品平行配制2 份,每份重復測定2 次。結果樣品中鹽酸羥考酮的含量分別為100.03%,100.82%,100.11%(n=4)。
3 討論
3.1 色譜條件選擇
緩釋制劑活性成分難提取[9-10],輔料有一定干擾。本品為強有機堿鹽酸鹽,選擇以離子對試劑為流動相的反相液相色譜系統進行試驗,根據各國藥典標準含量測定方法[11-14],擬訂了初步的含量測定色譜條件,即色譜柱為C18柱,流動相為1.1 g/L 1-庚烷磺酸鈉溶液(用50%磷酸溶液調pH至2.0)-乙腈-甲醇(738∶102∶160,V/V/V),流速為1.2 mL/min,柱溫為40 ℃,檢測波長為230 nm,進樣量為20μL。后續研究中發現,該方法存在輔料干擾主峰及活性成分難以提取的風險,樣品含量偏低(95.62%~97.64%)。降低流動相中有機相的比例,將主峰保留時間延后,輔料峰保留時間未發生明顯變化,兩色譜峰有分離趨勢,表明主峰對流動相的改變更敏感;提高甲醇比例,輔料峰與主峰完全分離,且出峰時間合適。故最終確定2.1項下色譜條件。
3.2 提取方法選擇
前期試驗中,考察研磨方法、濾液棄去體積量、提取方法(超聲及振搖)、溶劑體積對提取效果的影響。發現提高研磨效率可升高提取效率;棄去不同體積量,結果無顯著差異,濾膜對樣品無吸附作用;超聲提取效率顯著高于振搖提取,超聲時間越長,提取越充分,選擇較大的提取體積可規避提取不完全的風險。取同一批樣品,按2.2項下方法制備供試品溶液,結果鹽酸羥考酮含量為100.72%,故最終確定2.2項下供試品溶液制備方法。
3.3 方法評價
本研究中建立的方法專屬性強、耐用性好、回收率高,可用于鹽酸羥考酮緩釋片中鹽酸羥考酮的含量測定。
