分子骨架編輯在藥物合成中的應用與展望
2023-12-11 11:23:04賀靜遠王澤鎧戚顯瑋陳開元卞江
大學化學 2023年10期
賀靜遠,王澤鎧,戚顯瑋,陳開元,卞江,*
1 北京大學化學與分子工程學院,北京 100871
2 北京大學物理學院,北京 100871
在有機合成領域,分子編輯的歷史貫穿始終。各種分子的后期修飾都屬于廣義的分子編輯,但經典的分子編輯往往是“外圍編輯”(Peripheral Editing),如過渡金屬催化的C—H鍵活化等。“骨架編輯”(Skeletal Editing)是近幾年[1]興起的全新領域,包括原子的插入、刪除與替換(圖1A)。
研究人員在1881年發現了碳原子插入的反應Ciamician-Dennstedt重排,這是一個吡咯和鹵仿在強堿的作用下發生擴環、生成3-鹵代吡啶的反應[2],反應機理如圖1B所示。該反應也可用來由吲哚合成3-鹵代喹啉。
后期,陸續出現了一些經典的基礎有機反應如Baeyer-Villiger反應(插入氧原子),Favorskii重排(刪除碳原子縮環),Wolff-Kishner-Huangminlon還原(刪除羰基氧原子),構成了分子編輯的雛形(圖1C),這些反應可便捷地實現一個原子的插入刪除,但大多條件劇烈,官能團兼容性差[3]。近年來,該領域又有了新進展,誕生了一批分子骨架編輯反應,成功突破了反應條件劇烈、選擇性差等問題[4-9]。在眾多骨架編輯反應中,發展較成熟的有四類反應:碳原子的插入與刪除、氮原子的插入與刪除(圖2A-D)[2,4-7]。其中,碳原子的插入反應與氮原子的刪除反應條件溫和、官能團兼容性好、副產物少,在天然產物與藥物的合成中有著廣泛的應用[10-13]。下面兩例分別講述這兩類反應在上述領域中的應用。
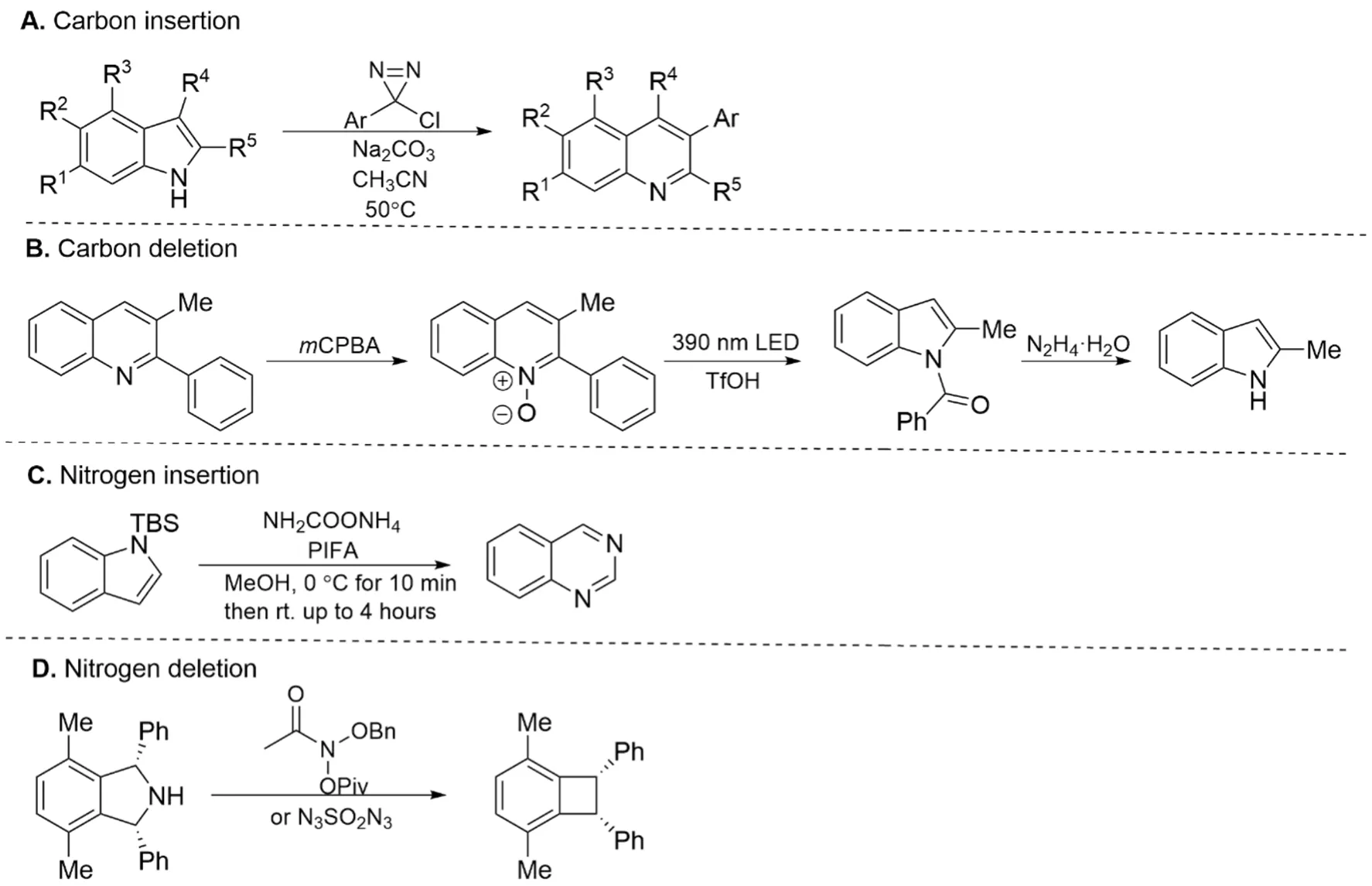
圖2 常見的分子編輯反應[2,4-7]
1 分子骨架編輯在天然產物與藥物全合成中的應用舉例
1.1 碳原子插入反應在Complanadine A全合成中的應用
Complanadine A是石松類生物堿Lycodine的不對稱二聚體,其四環骨架與吡啶環的引入一直是有機合成工作者們的工作熱點[14]。
關于吡啶環的合成思路有很多,比較著名的有以羰基化合物為原料、運用縮合反應進行合成的Hantzsch合成法、Kr?hnke合成法,此外還有環加成法(分別對應圖3A-C)[15-17]。Sarpong課題組設計的Complanadine A的合成路線比較傳統,采用連續的氧化、三氟甲磺酰化、鈀催化還原偶聯進行吡啶環的合成[15](圖4A)。該方法從羰基化合物出發,原料易得、操作方便,且可以以較高的產率得到吡啶衍生物,但是由于Complanadine A中存在3-取代基(圖4C中4n結構中左側吡啶環以氮原子為1號,順時針編號,下同),吡啶的2位與4位較為活潑,而對吡啶環進行3位活化非常困難[18]。該策略后續對吡啶環3位的活化使用了大量的步驟,使合成路線變得繁瑣。

圖3 吡啶的傳統合成方法[15-17]
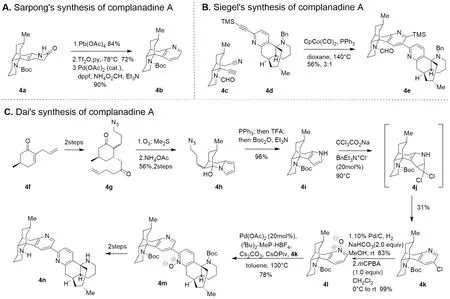
圖4 Complanadine A中關鍵吡啶環的三種合成思路[11,14,18]
Seigel課題組巧妙設計底物,通過連續的[2+2+2]環加成反應巧妙而高效地構建了Complanadine A的二聚體骨架,有效避免了吡啶3位活化困難的問題[18],但是前期改造設計底物也使得合成路線步驟繁瑣。同時底物之一腈的活性太弱,反應需要較為劇烈的條件,并且該合成思路過于巧妙,并不普遍,無法形成易于遷移運用的合成范式(圖4B)。
而Dai課題組采用分子編輯的方法,先在環中引入富電子的吡咯環,利用其親核性進行縮合構建四環骨架,而后利用基于Ciamician-Dennstedt重排的碳原子插入反應插入一個氯代卡賓,不僅插入了一個碳原子擴環為吡啶,也同時實現了吡啶3位的活化[11](圖4C)。該方法有效解決了吡啶類化合物合成中的兩個大問題,其一是構筑吡啶環的同時構建復雜的分子骨架,其二是吡啶3號碳原子的活化,由此可見分子編輯中的碳原子插入法為吡啶類分子的合成提供了全新的、可以形成一種固定模式的思路,在3號碳修飾的瓶頸問題上也做出了突破。
1.2 氮原子刪除在Piperarborenine B全合成中的應用
含有環丁烷結構的生物堿往往具有顯著的抗癌、抗菌等生物活性,Piperarborenine家族的化合物便是其中之一[19]。Piperarborenine是從胡椒中提取出的一系列生物堿,包括Piperarborenine A-D,它們都是多取代的環丁烷衍生物。
多取代環丁烷在合成上的難度主要在于環丁烷四元環自身的不穩定性導致在修飾的同時很難保證環丁烷骨架完好,同時在環丁烷骨架上進行取代的方法也非常有限,目前主要局限于過渡金屬催化的偶聯[20],這類反應的產率在40%-80%之間,盡管產率不算低,但像這樣依次添加取代基,會導致合成路線較長且產率會大打折扣,這為合成造成了很多困難。
傳統的環丁烷骨架合成方式是[2+2]環加成反應,圖5A中Tang課題組的合成路線就是一個代表[21]。這種方法的主要問題在于產物的立體選擇性與區域選擇性難以控制,會得到各種[2+2]環加成反應的產物。盡管近年來發展出了高立體選擇性的[2+2]環加成反應,如Tang課題組的方式,但后續在環丁烷骨架上插入取代基的步驟不僅繁瑣,而且產率低。并且[2+2]反應若要實現盡可能高的選擇性,其對底物烯烴取代基的電子效應有著嚴格的要求,使得底物的選擇大大受限。
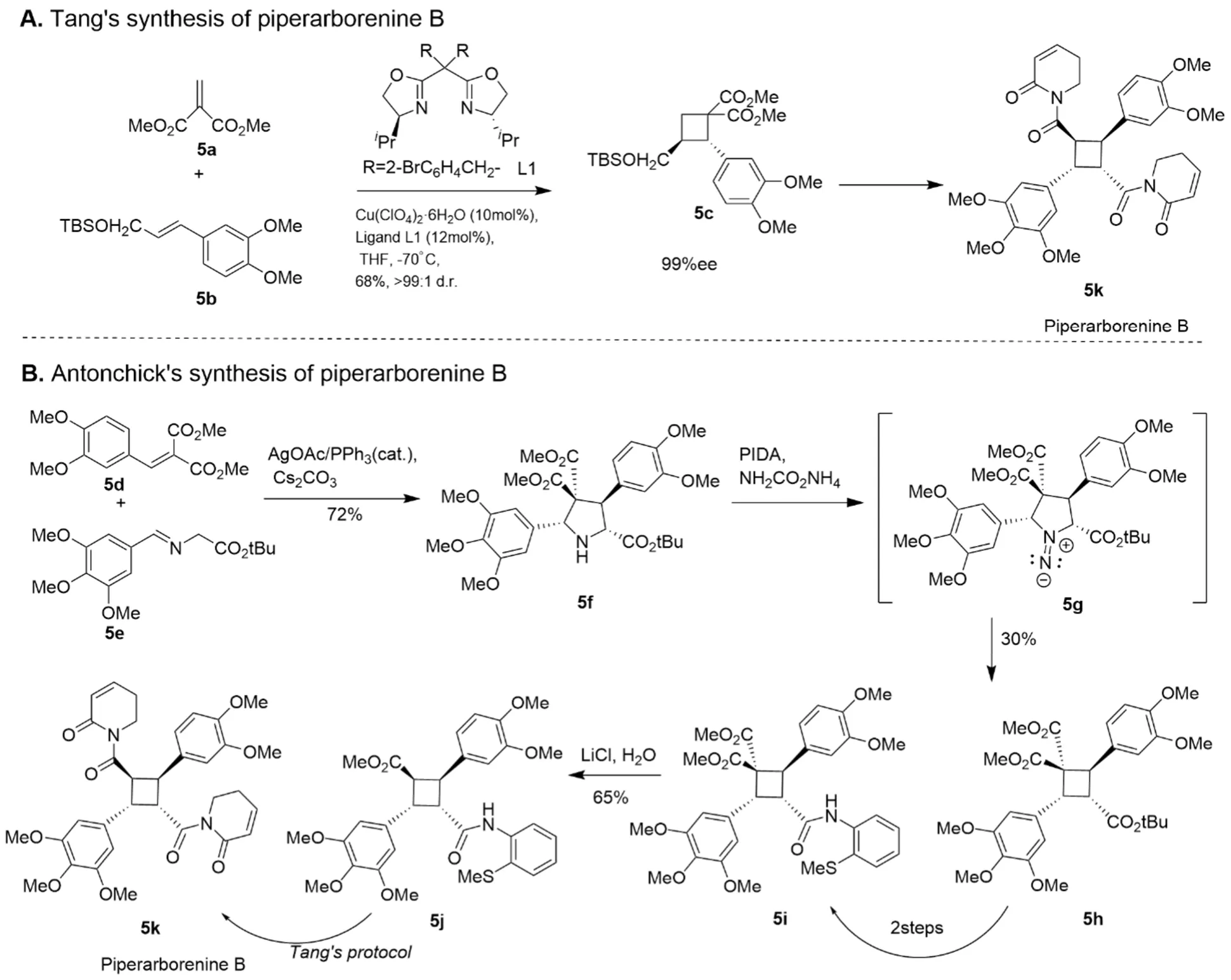
圖5 Piperarborenine B的兩種合成思路[10,21]
而Antonchick課題組利用分子編輯方法,利用Schiff堿衍生物與烯烴發生[3+2]環加成反應得到四氫吡咯骨架,而后利用PIDA (碘苯二乙酯,一種高價碘試劑)與氨基甲酸銨氮源原位生成氮賓,實現氮原子刪除,得到環丁烷骨架[10](圖5B)。這種四氫吡咯策略的優點主要在于:第一,[3+2]反應的選擇性較高,遠好于[2+2]反應;第二,底物之一Schiff堿的合成方法成熟多樣,相比于取代烯烴而言更容易獲得;第三,逆合成分析中,氮原子有四個位點可以插入,這意味著理論上至少有4組不同的底物組合可供選擇,這拓展了底物的選擇范圍;第四,四氫吡咯骨架穩定,且在該骨架上進行取代基修飾的方法多樣,可以便捷高效地得到目標產物。運用分子編輯法可以轉換目標骨架,使用更加合適的起始原料進行更簡潔明快的合成。
2 前沿進展——原子替換反應及其應用
骨架編輯在全合成中初露頭角,但前面提到由于替換反應發展不成熟,這大大限制了它的廣泛應用。近兩年在骨架編輯領域的新進展則開發了兩例原子替換反應,彌補了這一領域的空白。
2.1 硼原子插入介導的氧到氮原子替換
1984年,Pachaly和West報道了一類硼烯對四氫呋喃C—O鍵的插入反應,實現了金屬對飽和醚鍵的選擇性插入[22]。2021年,Lyu課題組發展了這一反應,形成了豐富的硼原子分子編輯方法學,不僅可以實現硼原子的插入,還可以以此為基礎,實現其他原子的插入與替換,是一類有效的醚類分子編輯方法[8](圖6A)。
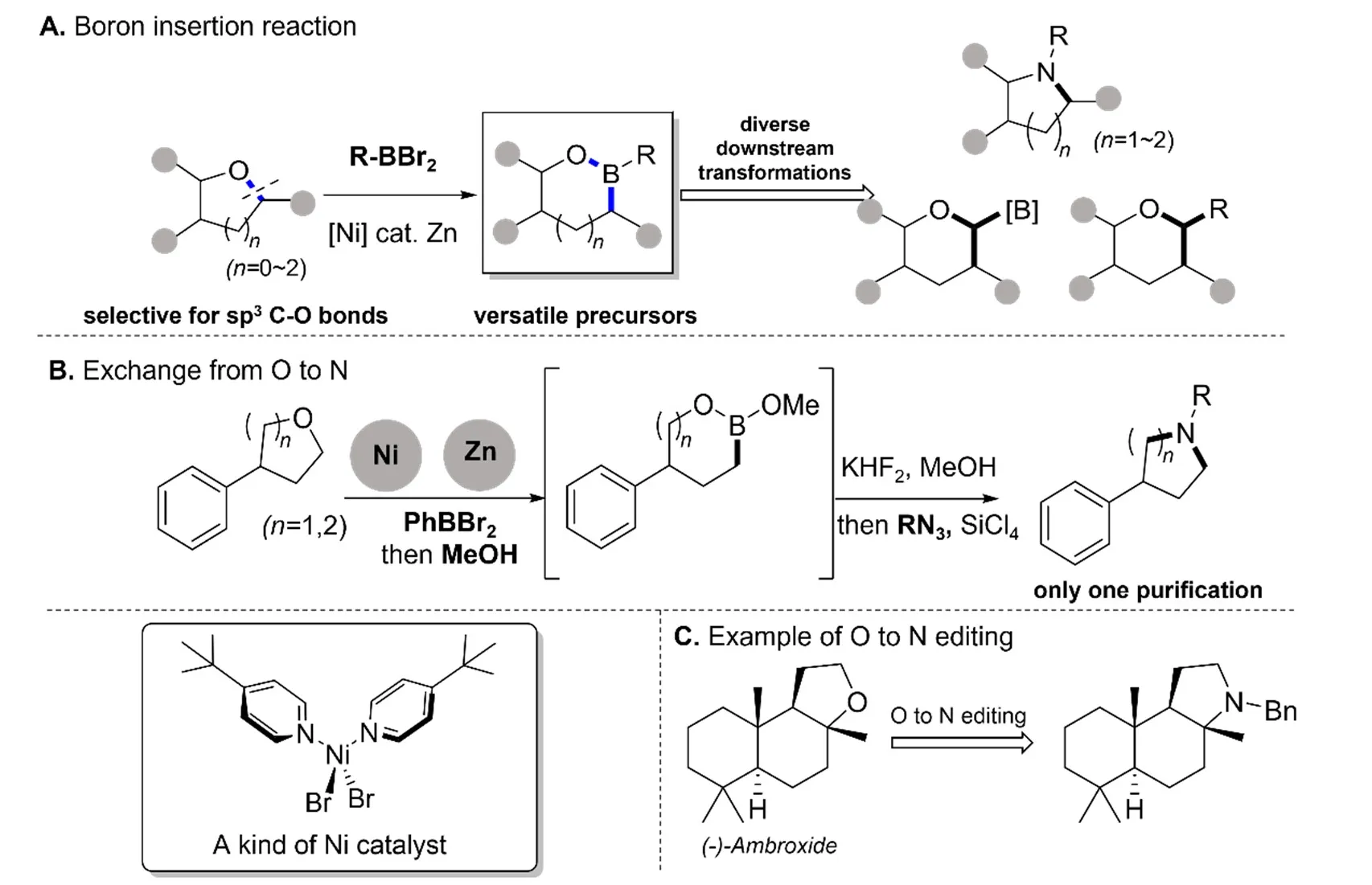
圖6 硼原子插入介導的氧到氮原子替換[8]
采用圖6B的條件可以實現環醚中氧原子到氮原子的替換[8],該反應選擇性高,主要受空間位阻的控制,并且底物兼容性好,適用于含有各種復雜或不穩定取代基的醚,且對于四氫呋喃衍生物的反應性尤其好。
同時該課題組將此反應應用到天然產物降龍涎香,通過三個步驟完成了骨架上氧到氮原子替換(圖6C),這為該反應在藥物合成中的應用提供了參考[8]。
2.2 碳氮原子替換
天然產物中有大量含有芳環結構的分子,如果能實現芳環骨架上原子的替換,這將為天然產物的合成、性質研究帶來極大的方便。2017年Pennington課題組就曾考慮過完成芳環中碳原子到氮原子的轉化[23],并將其應用到藥物設計與合成領域(圖7A(a)),因為這可以大大改善藥物性能。有趣的是,由N原子到C原子的替換也曾被提出應用[24](圖7A(b))。圖7A(b)中化合物7c是一種藥物合成前體,對PARP酶有一定的抑制作用(Ki= 5.8 nmol·L-1),運用分子骨架編輯的思路將一個氮原子替換為碳原子,便得到了抗癌藥物魯卡帕尼7d,其對PARP酶的抑制作用大大增強(Ki= 1.4 nmol·L-1)。

圖7 碳氮原子的替換反應及其在藥物設計中的應用[23-26]
由于氮原子作為雜原子在芳環中的活潑性,由氮原子向碳原子的轉化相對簡單,例如2007年Fout課題組報道的鈦試劑對雜環的脫氮研究,完成了該轉化[25](圖7B(a));以及2021年Morofuji課題組報道的將對取代吡啶轉化為偏取代苯胺的反應等[26](圖7B(b))。
芳環中由碳原子到氮原子的替換則要晚一些。2022年10月Patel課題組開發了由苯環到吡啶的碳原子替換反應[9],該反應由氮原子插入、碳原子刪除兩步組成,以芳基疊氮為底物,光氧化破壞芳香性插入氮原子,后通過6π電環化刪除碳原子得到2-氨基吡啶,作者提出的可能反應機理如圖8所示。這一反應可以實現一鍋煮且兼容多種復雜官能團。
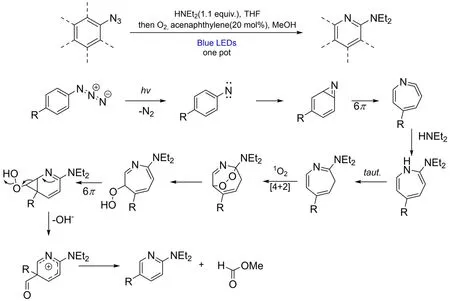
圖8 將碳原子替換為氮原子,實現苯環到吡啶的轉化,反應式以及作者提出的可能的反應機理[9]
3 討論與展望
結合上述案例,相較傳統的有機分子合成,我們可以總結分子骨架編輯的幾大優點。首先,其立體選擇性與區域選擇性好,可以用來合成一些手性分子,且方法簡單條件溫和。其次,它們可以簡化逆合成分析,為復雜分子提供了更加便捷的合成路線。再者,其官能團的兼容性好,當骨架上有很多復雜且敏感的取代基時,可以做到只改變骨架而不影響官能團。因此,分子骨架編輯的手段在多個領域具有獨特的優勢。
3.1 分子編輯與藥物化學
分子編輯在藥物化學領域有著極為廣闊的應用前景,這主要表現在藥物的合成與優化。
藥物分子結構復雜,每合成一種新藥,幾乎就意味著要從頭設計合成路線。但事實上很多藥物分子都是從同一種結構衍生出來的,因此在藥物合成中,人們將藥物分子拆成一個個合成砌塊(Building Blocks),通過構建各樣的合成砌塊進行分子庫合成[27],以減少對每一種分子從頭設計合成路線的麻煩,較為方便地合成出大量不同的藥物分子。2022年諾貝爾化學獎的成果點擊化學便是分子庫合成的重要手段——如構建含有炔基和疊氮的合成砌塊[28],通過高效的環加成反應進行高通量的分子庫合成。但點擊化學中大部分底物與產物結構較為特殊,所以能通過它實現的分子庫合成較為有限,而分子編輯則拓寬了分子庫合成的邊界,減少了新藥合成的工作量。
比如,圖9A中結構9a是一些商業藥物如9b-9d結構里的通式,為了合成一系列含有結構9a的分子,研究人員采用氮原子刪除反應作為分子庫合成的手段。以含有氨基和羰基的小分子(合成砌塊多來自于藥物設計常用的結構) 9ea-9ef、9fa-9ff作為合成砌塊,通過縮合反應得到二級胺,最后通過氮原子刪除反應構建碳碳鍵,得到候選分子9ga-9gf[27]。這樣,只需要沿著一個思路,分子骨架編輯就可以幫助人們合成各種各樣的藥物分子(圖9A)。
以分子骨架編輯為主要技術的分子庫合成不僅在藥物合成中嶄露頭角,在藥物優化上也有較高的應用價值。在藥物設計領域,一種新藥的發明意味著此后需要進行大量的嘗試以改變藥物分子的結構,進行藥理學實驗,尋找藥效更優的分子結構。在過去若想實現這一工作,幾乎都要從頭設計新的合成路線,這樣的工作量無疑是驚人的,也限制了藥物優化的進展。而分子庫合成可以只改變合成砌塊,通過排列組合合成出大量不同結構的藥物候選分子,從而便捷實現了藥物的高通量篩選。
在藥物優化工作中,除了構建合成砌塊進行分子庫合成,直接對分子骨架進行后期編輯也是一種可行的策略。因為候選分子與原始藥物的差別往往僅是一個骨架原子,這正是分子骨架編輯擅長實現的轉化[4](圖9B)。例如,由于氮原子的數目對藥物分子的極性、pKa、穩定性等性質影響很大,因此在藥物分子的骨架中增刪氮原子是藥物設計優化的主要方向之一。采用分子編輯,理論上可以通過一步反應便捷地增刪氮原子,很大程度上簡化了藥物的優化工作。吡啶具有“必需氮原子效應”(Necessary Nitrogen Atom Effect)[9],其對藥物性質有很大影響,因此將苯環替換為吡啶是藥物優化工作的重要方向之一。例如2.2節提到的一種Cdc7激酶抑制劑的優化便采用了這一思路,將苯環替換為吡啶,使得IC50值顯著下降,顯著增強了藥效[23](圖7A(a))。但目前該領域發展尚不成熟,以猜想與理論預測居多,實驗成果較少,從理論到真正的藥物優化仍有較長的路要走。
除了對藥物中氮原子進行編輯,對碳原子、氧原子的編輯在藥物優化中也有應用。碳原子數目影響著碳環的大小,進而影響藥物的穩定性、藥效持續時長、降解速度;氧原子影響著分子的極性,同時能參與氫鍵的形成,改變分子的水溶性等性質。
3.2 分子編輯在其他領域的應用與展望
除了藥物領域,分子編輯在天然產物全合成、AI輔助研究中亦前景廣闊。正如第1節提到的,分子編輯可以構建新型的有機合成模式,在復雜骨架、不穩定骨架的合成中展現出極大的優勢,可以實現無保護基合成以及復雜骨架的高效合成。
我們認為,分子編輯在未來最引人注目的應用應當是AI輔助進行有機合成。傳統的合成路線是前人智慧的結晶,這一類合成路線大多巧妙,只適用于某一特定分子的合成,難以形成模式化的合成思路,不容易被機器學習。而分子編輯由于其直接、簡潔的特點,易于被機器學習并形成范式,這將促進AI在有機合成領域的廣泛應用。
3.3 分子編輯的局限性
當然,分子骨架編輯也有一定的局限性。這類反應的底物很局限,僅適用于某一類底物,比如C、N原子的插入只適用于像吲哚一類的富電子芳環[2,5],C原子的刪除只適用于喹啉,N原子的刪除只有二級胺發展較為成熟,而原子的替換在目前對于芳環幾乎難以實現[8](圖10)。
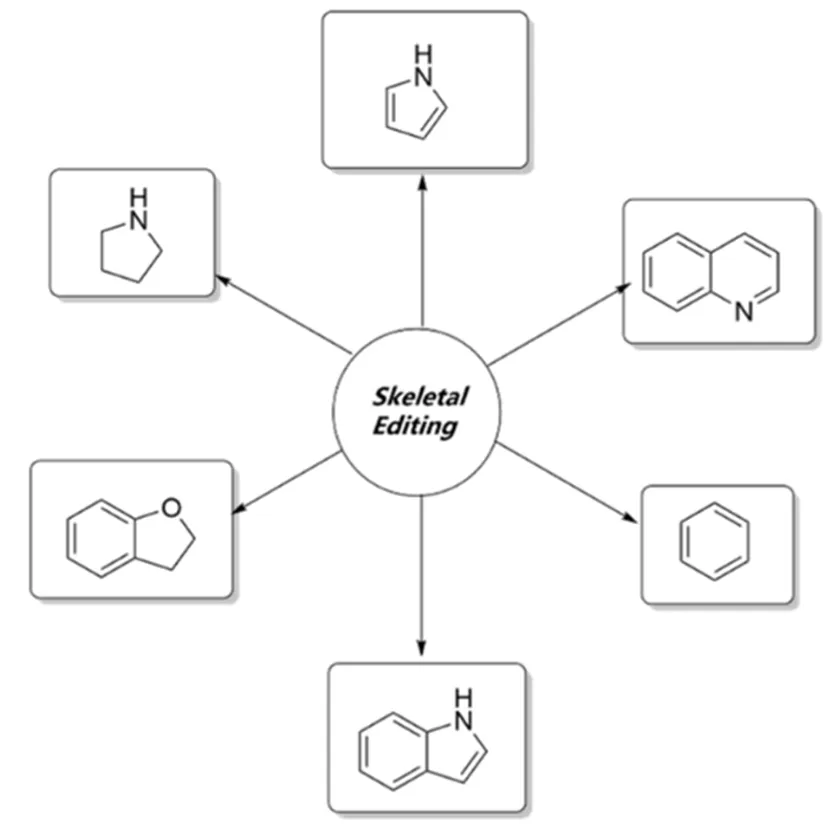
圖10 目前分子骨架編輯常用的底物,依然較為局限[2,5,8]
不過,這種局限性僅能說明目前分子骨架編輯方法不夠成熟完善,有待完整體系的構建。我們相信,隨著分子編輯體系日益完備,其定能引發有機合成領域的一場革新。總而言之,分子骨架編輯的方法是充滿未知的,擁有廣闊前景的,值得被新一代化學研究者學習和探索。
