高效液相色譜三重四極桿質譜聯用法測定茶葉中百草枯殘留量
2023-09-05 04:21:04汪文俊徐小雷余曉峰
現代食品 2023年13期
關鍵詞:方法
◎ 吳 瓊,汪文俊,朱 林,徐小雷,宗 凱,韓 芳,葉 菁,余曉峰
(1.合肥海關技術中心,食品安全分析與檢測安徽省重點實驗室,安徽 合肥 230022;2.黃山市茶產業促進中心,安徽 黃山 245000)
百草枯是一種快速滅生性除草劑,能夠迅速被植物綠色組織吸收,可有效殺死一年或多年生雜草,在土壤中迅速與土壤結合而使其鈍化[1-3]。因此,它們曾長期被認為是對環境無害類農藥而在全球推廣使用,但百草枯對人和動物毒性極大,一旦誤服無藥可解,口服中毒死亡率在90%以上,我國是百草枯生產使用大國,也是中毒者數量最多的國家[4]。《食品安全國家標準 食品中百草枯等43 種農藥最大殘留限量》(GB 2763.1 2018)發布后,食品安全國家標準增加了百草枯在谷物和茶葉中的最大殘留限量要求,目前現行的《食品安全國家標準 食品中農藥最大殘留限量》(GB 2763 2021)延續了百草枯在茶葉中的最大殘留限量要求(0.2 mg·kg-1)。國外標準中,歐盟對輸歐茶葉中百草枯殘留量作了規定,不得大于0.05mg·kg-1。
目前現行的百草枯檢測標準有《出口植物源性食品中百草枯和敵草快殘留量的測定 液相色譜-質譜/質譜法》(SN/T 0293 2014)、《出口果蔬中百草枯檢測拉曼光譜法》(SN/T 4698 2016)和《稻米中百草枯殘留量的測定 液相色譜-質譜/質譜法》(DB22/T 1622 2012),檢測方法主要包含液相色譜串聯質譜法和拉曼光譜法等,檢測對象涉及水果蔬菜、玉米、稻米等植物源性食品。SN/T 0293 2014作為GB 2763 2021 指定的百草枯檢測方法,其適用范圍僅為大米、大豆、玉米、小麥、棉籽、干木耳、甘藍、蘋果、香蕉和草莓,未包含茶葉。直接使用該方法對茶葉中百草枯殘留量進行檢測時存在溶劑用量大且回收率偏低的情況。覃重陽等[5]利用表面增強拉曼光譜法對茶葉中百草枯農藥殘留進行快速檢測,但該方法在分析不同茶葉中農藥含量時存在基質影響大的問題,普適性較差。目前主流的百草枯殘留量檢測方法仍為高效液相色譜質譜聯用法[6-15]。色譜質譜聯用技術具有選擇性好、定性能力強、靈敏度高的優點,近年來較少有該方法對茶葉中百草枯殘留量的檢測研究報道。
本研究以茶葉為檢測對象,在《出口植物源性食品中百草枯和敵草快殘留量的測定 液相色譜-質譜/質譜法》(SN/T 0293 2014)方法基礎上,采用固相萃取結合高效液相色譜三重四極桿質譜聯用技術,通過優化前處理過程及儀器條件等,建立了一種準確、高效測定茶葉中百草枯殘留量的方法。本方法定量限為5 μg·kg-1,可滿足國標和歐盟對茶葉的限量要求,為完善百草枯檢測標準、監控茶葉質量安全和國家食品安全抽檢提供了技術支撐。
1 材料與方法
1.1 材料與試劑
本實驗中所用綠茶、紅茶、黑茶等茶葉樣品均購自黃山茶城。
甲醇、乙腈(色譜純,西隴科學);甲酸(色譜純,阿拉丁);氨水(分析純,阿拉丁);超純水由純水儀制得(Milli-Q,美國Millipore);WCX 固相萃取柱(60 mg,3 mL,美國Waters);百草枯二氯酸鹽(CAS:1910-42-5,100 mg·L-1,天津阿爾塔)。
1.2 儀器與設備
樣品磨(KN295,丹麥FOSS);渦旋振蕩儀(Vortex-Genie 2,美國Scientific Industries);分析天平(萬分之一,上海梅特勒);固相萃取裝置(美國Waters);液相色譜-質譜聯用儀(1290II-6470,美國安捷倫)。
1.3 實驗方法
1.3.1 標準溶液配制
標準儲備液:移取1 mL 質量濃度為100 mg·L-1的百草枯二氯鹽有證標準儲備液于100 mL 容量瓶中,用乙腈定容至刻度,得到1 mg·L-1百草枯二氯鹽標準儲備液。
標準工作溶液:移取相應體積的標準儲備液(1 mg·L-1)用空白樣品基質溶液稀釋,分別配制成質量濃度為5.0 μg·L-1、10.0 μg·L-1、20.0 μg·L-1、50.0 μg·L-1、100.0 μg·L-1和200.0 μg·L-1的系列標準工作溶液。
1.3.2 樣品前處理
取代表性的茶葉樣品500 g 經樣品磨粉碎后充分混勻。稱取2.0 g 樣品于50 mL 具塞離心管中,加入20 mL 含5% FA 的甲醇水溶液,渦旋混勻3 min 后均質提取2 min,4 000 r·min-1離心5 min,取上層提取液10 mL 到15 mL 聚乙烯離心管中,用5% NH4OH 水溶液調節pH 值至7.0 0.5,將上清液轉移至已經過活化的固相萃取柱內,控制流速,棄去流出液。依次用2 mL 水和2 mL 甲醇淋洗萃取柱,最后用4 mL 含10% FA 的乙腈溶液洗脫,控制流速,收集洗脫液于刻度離心管中,洗脫液經45 ℃氮吹至干,取1 mL 乙腈-0.1% FA 水溶液溶解殘渣,過0.22 μm 濾膜后,供液相色譜三重四極桿質譜聯用儀測定。
1.3.3 色譜條件
色譜柱:Thermo Syncronis HILIC(長度為100 mm,直徑為2.1 mm, 粒徑為1.7 μm); 流動相A:0.1% FA 水溶液;流動相B:乙腈;梯度洗脫程序:0 ~3.0 min,5% A,3.0 ~5.0 min,5% ~90%A;5.0 ~8.0 min,90% A;8.0 ~8.1 min,90%~5%A;柱溫:40 ℃;進樣量:2 μL;流速:0.3 mL·min-1。
1.3.4 質譜條件
離子化模式:電噴霧電離正離子模式(ESI+);質譜掃描方式:多反應監測(MRM);干燥氣:氮氣;鞘氣流速:10 L·min-1;輔助氣流速:3 L·min-1;離子傳輸管溫度:350 ℃;噴霧氣電壓:4 000 V;碰撞氣:氬氣,1.0 mTorr,質譜參數見表1。

表1 百草枯的質譜條件表
1.4 數據處理
采用高效液相色譜質譜聯用儀自帶軟件工作站[Agilent Mass Hunter Quantitative Analysis(for QQQ)]進行標準曲線繪制和樣液質量濃度計算。采用Microsoft Office Excel 2013 軟件進行數據統計及統計圖表繪制,采用Win 10 自帶畫圖軟件進行圖像輔助處理。
2 結果與分析
2.1 色譜與質譜條件的優化
接雙通注入質量濃度為1 mg·L-1的百草枯標準溶液以優化質譜條件,通過全掃描模式(Q3 SCAN)優化噴霧氣電壓、源內碎裂電壓、干燥氣流速和離子傳輸管溫度等參數,得到對應母離子186;再通過產物離子掃描模式(Product ion SCAN)找到對應子離子;分別選擇響應較優的兩個產物離子作為定量和定性離子,分別為171 和155;最后在多反應監測(Multiple Reaction Monitoring,MRM)模式下根據響應優化碰撞能等參數[4],具體參數見表1。
百草枯為親水強極性化合物,參考SN/T 0293 2014 方法采用親水性Syncronis HILIC 色譜柱進行分離。流動相采用0.1% FA 水—乙腈流動相體系優化峰形減少拖尾,同時提高離子化效率,利用優化好的質譜參數和色譜條件進行MRM 分析,百草枯(0.2 mg·L-1)的總離子流圖與MRM 質譜圖見圖1。
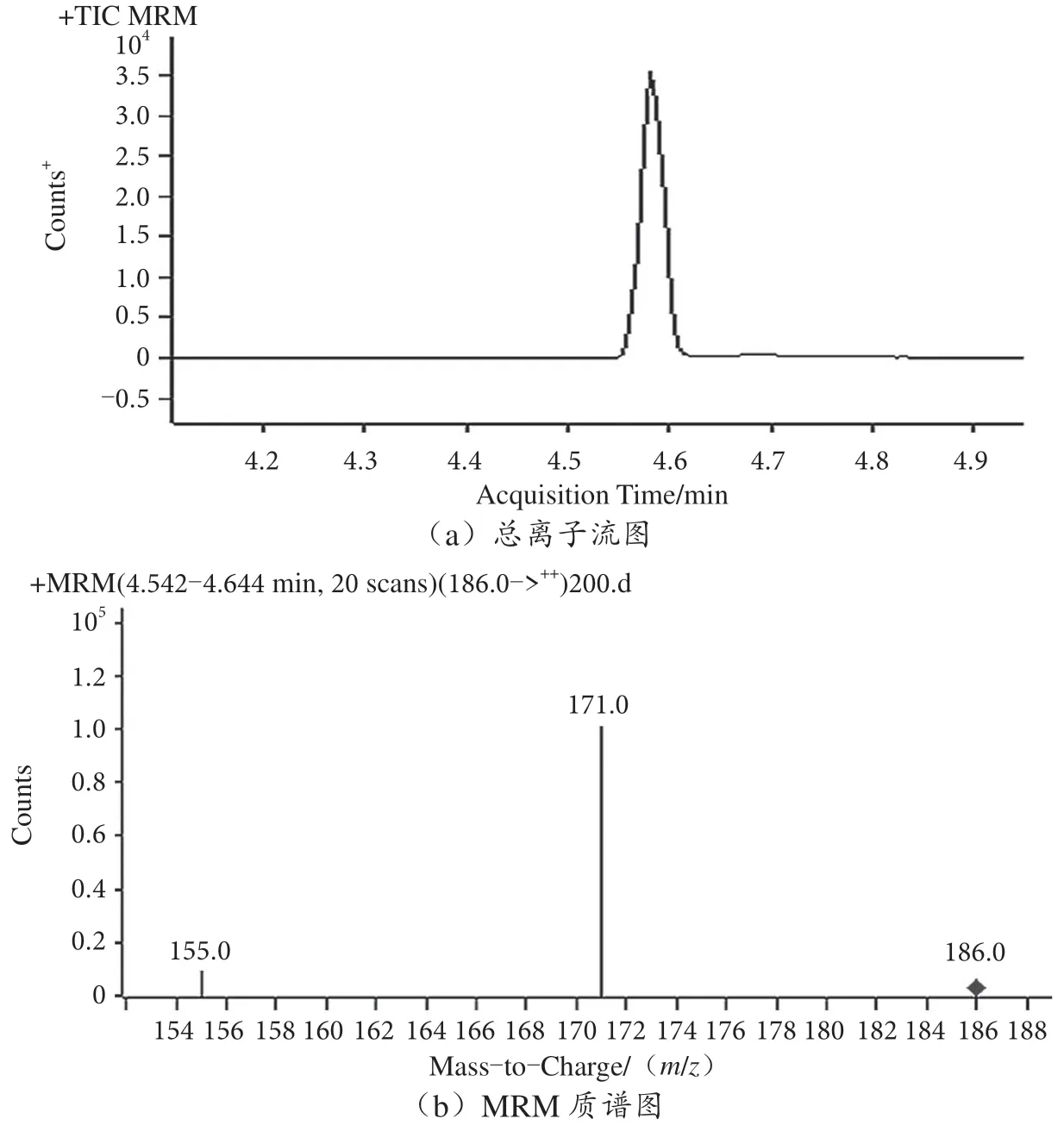
圖1 百草枯(0.2 mg·L-1)總離子流圖與MRM 質譜圖
2.2 提取溶劑的優化
百草枯呈堿性,易溶于水,在中性和酸性條件下穩定。調研文獻[6-7,11]發現,大部分采用有機溶劑與稀酸混合溶液進行提取,分別選擇相同體積甲醇/0.1 mol·L-1鹽酸溶液(1 ∶9)、乙腈/0.1 mol·L-1鹽酸溶液(1 ∶9)、含5% FA 的甲醇/水(1 ∶1)、含5% FA 的乙腈/水(1 ∶1)進行溶劑萃取,考察實際茶葉樣品加標的萃取效率。實驗發現,選擇含5%FA 的甲醇/水(1 ∶1)溶液萃取效率最佳,百草枯歸一化萃取效率見圖2。
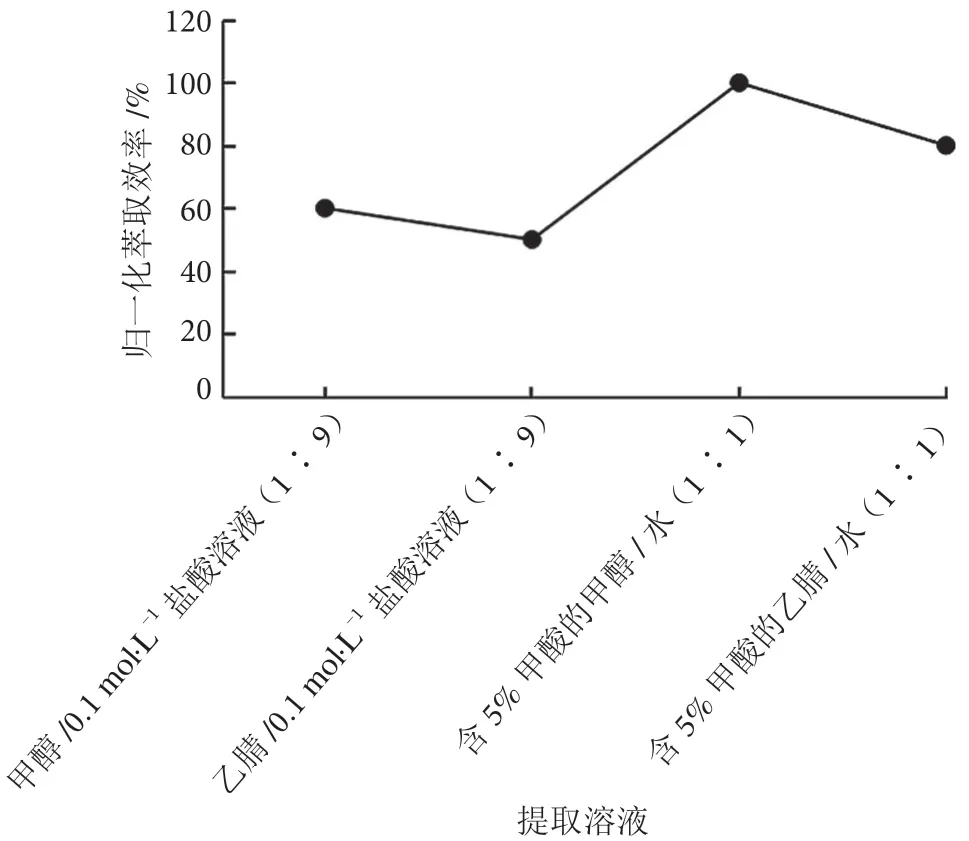
圖2 不同提取溶劑歸一化萃取效率圖
2.3 固相萃取柱洗脫條件的優化
茶葉樣品基質較蔬菜水果復雜,提取液色素含量高,基質效應強,無法直接上機分析。為了獲得更好的凈化效果,采用固相萃取法凈化。百草枯為親水性強極性化合物,參考SN/T 0293 2014 方法,選用陽離子交換、混合型WCX 固相萃取柱進行凈化。洗脫條件是決定回收率的關鍵因素,在0.2 mg·kg-1加標濃度下,分別用含5%、10%、15% FA 的乙腈溶液作為洗脫液,進行回收率實驗。實驗發現,含10%、15%FA 的乙腈溶液的洗脫效率明顯優于5%,而用10%和15% FA-乙腈時的洗脫效率相差不大,考慮到相同洗脫效率下優先選擇含酸濃度低的乙腈溶液作為洗脫液,最終采用含10% FA 的乙腈進行洗脫。不同洗脫溶劑歸一化洗脫效率結果見圖3。同時,實驗考察了不同體積(2 mL、3 mL、4 mL 和5 mL)洗脫液的洗脫效率。在0.2 mg·kg-1的加標濃度下,根據回收率結果最終選用4 mL 洗脫液洗脫。不同洗脫體積下的回收率結果見圖4。
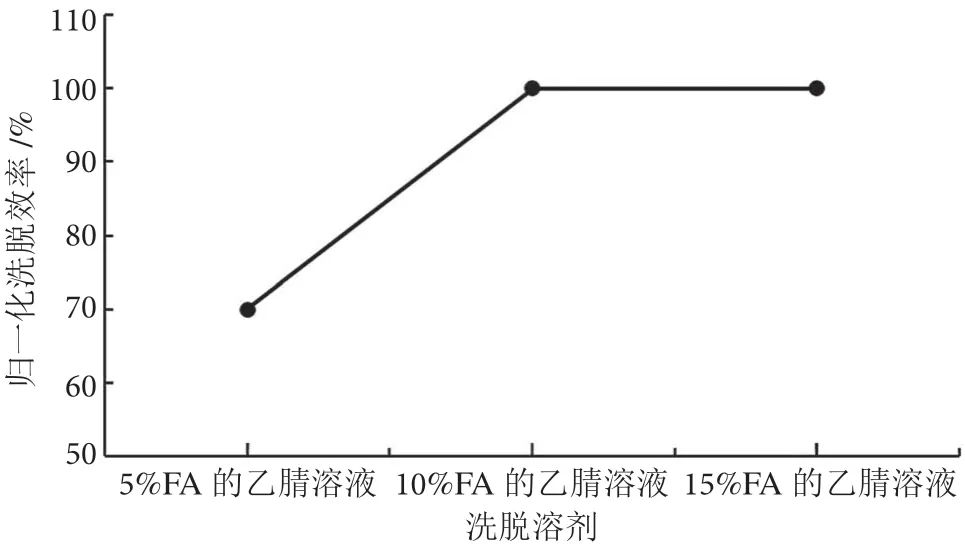
圖3 不同洗脫溶劑歸一化洗脫效率圖
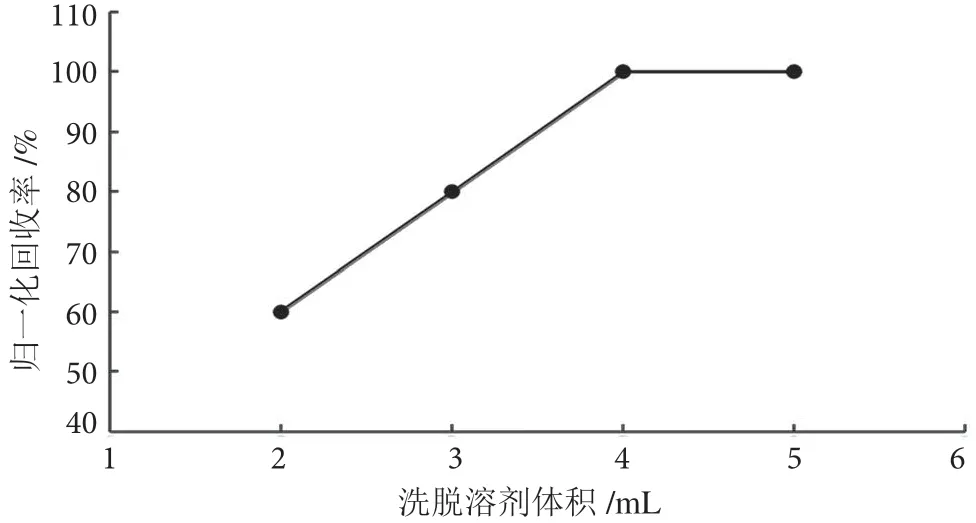
圖4 不同體積洗脫液洗脫歸一化回收率圖
2.4 線性關系、方法定量限、回收率和精密度實驗
用空白基質溶液配制質量濃度為5.0 ~200.0 μg·L-1的百草枯系列標準工作溶液,1.3 色譜和質譜條件下進樣,以百草枯定量離子171 色譜峰面積為y軸,質量濃度為x軸繪制標準曲線。結果表明,在相應質量濃度范圍內線性良好,線性方程為y=307.51x-480.63,r2=0.999 86。
以3 倍信噪比確定檢出限,該方法百草枯檢出限為1 μg·kg-1,以10 倍信噪比確定定量限,該方法百草枯定量限為5 μg·kg-1。
為了考察該方法在不同茶葉中的適用性,分別選擇綠茶、紅茶、黑茶陰性樣品,在方法定量限(0.005 mg·kg-1)、歐盟茶葉限量(0.050 mg·kg-1)、標曲最高點/國標茶葉限量(0.200 mg·kg-1)3 個不同濃度水平下加標,按照本研究方法進行處理和測定,每個加標水平做6 次平行實驗,加標回收率和精密度實驗數據如表2 所示。結果表示,百草枯平均回收率為92.5%~103.2%,相對標準偏差為3.2%~5.6%,準確度和精密度滿足方法學要求。

表2 陰性樣品中百草枯加標回收率和精密度表(n=6)
2.5 方法應用
用本文建立的方法對市售20 份茶葉樣品進行百草枯殘留量檢測,20 份樣品均無百草枯殘留檢出,市售樣品和陰性加標樣品MRM 圖見圖5 和圖6。
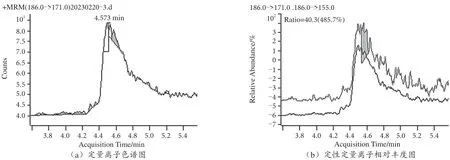
圖5 市售樣品MRM 圖
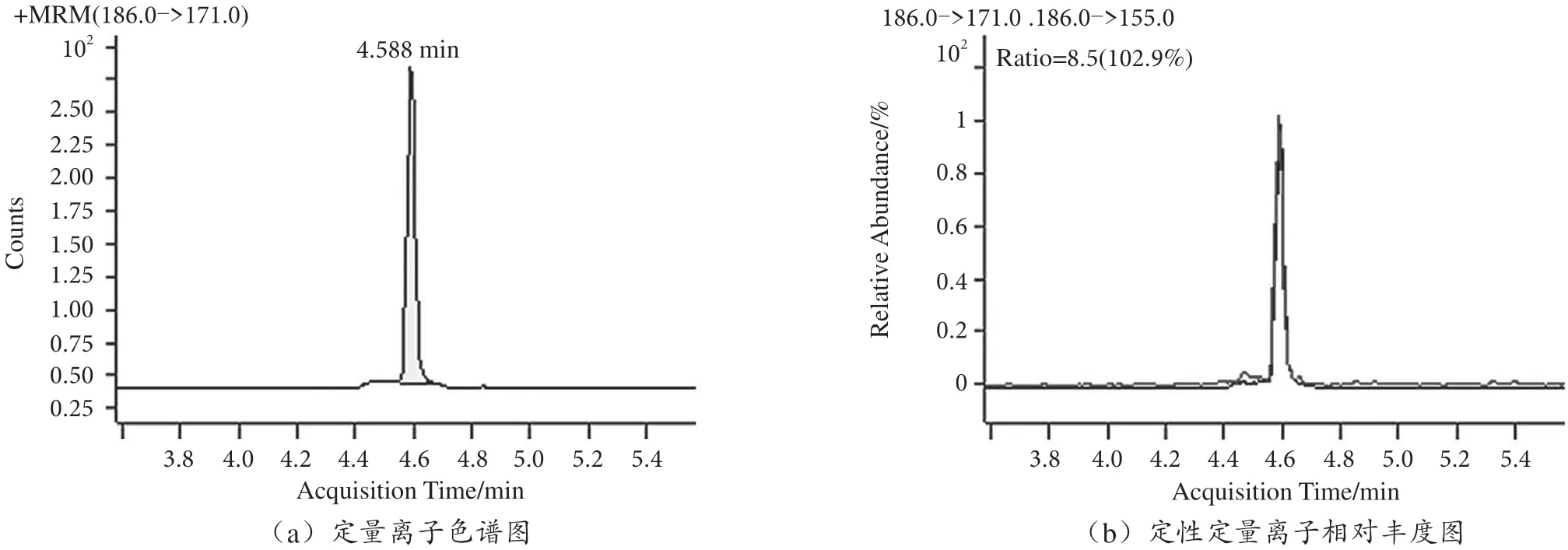
圖6 茶葉加標樣品MRM 圖
3 結論與討論
本文建立了一種茶葉中百草枯殘留量的固相萃取/高效液相色譜三重四極桿質譜檢測法,茶葉樣品經甲醇-甲酸水溶液提取后,采用WCX 固相萃取柱凈化,樣液在含0.1% FA 水-乙腈流動相體系經親水性HILIC 色譜柱分離,采用電噴霧離子源正離子模式采集,多反應監測模式下檢測,外標法定量。由于百草枯易產生柱殘留,在分析標準工作溶液與樣液之間可插入數針試劑空白,避免柱殘留產生的假陽性。該方法結果準確,適用于各種茶類中百草枯殘留量的測定,方法定量限可滿足各國對茶葉中百草枯的限量要求,為完善百草枯檢測標準、監控茶葉質量安全、國家食品安全抽檢提供了技術支撐。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
