分散固相萃取-UPLC-MS/MS法測定水蛭中10 種磺胺類藥物殘留
2023-07-07 00:45:54王小喬許曉輝張虹艷孫玉婧吳福祥潘秀麗
食品與藥品 2023年3期
王小喬,許曉輝*,張虹艷,馮 翀, ,孫玉婧,吳福祥,潘秀麗,李 赟
(1. 蘭州市食品藥品檢驗檢測研究院/國家市場監(jiān)管重點實驗室(食品中農(nóng)藥獸藥殘留監(jiān)控),甘肅 蘭州 730050;2. 蘭州理工大學 生命科學與工程學院,甘肅 蘭州 730050;3. 蘭州職業(yè)技術(shù)學院,甘肅 蘭州 730070)
中藥材水蛭是由水蛭科動物螞蟥、水蛭或柳葉螞蟥經(jīng)炮制而得的干燥品,具有破血、逐瘀、通經(jīng)的功效,臨床廣泛用于治療徽瘕、痞塊、血瘀閉經(jīng)、跌打損傷等癥[1-2]。隨著水蛭的需求量增加,傳統(tǒng)野生水蛭供應(yīng)已不能滿足市場需求。為了保護野生資源、化解供需矛盾,人工養(yǎng)殖逐漸成為水蛭的主要來源[3-5]。磺胺類藥物有抗菌高效、化學性質(zhì)穩(wěn)定、使用方便和價格便宜等優(yōu)點,被廣泛應(yīng)用于水產(chǎn)養(yǎng)殖業(yè)。目前,由于在水蛭養(yǎng)殖過程中有不規(guī)范或超量使用抗生素類獸藥的情況,導致獸藥在水蛭體內(nèi)不能完全代謝;且磺胺類藥物在人體內(nèi)蓄積后會產(chǎn)生細菌耐藥性,且有潛在的致癌性,不利于人體健康[6-8]。我國食品安全國家標準《GB 31650-2019 食品安全國家標準 食品中獸藥最大殘留限量》對動物源性食品中磺胺總量作了明確規(guī)定,動物源性食品中磺胺類藥物的最高殘留限量(MRL) 為 0.1 mg/kg[9]。當前,水蛭中磺胺類藥物殘留的檢測方法未見報道,因此,建立一種高效、便捷、準確檢測水蛭中磺胺類藥物殘留的方法具有重要的研究意義。
目前,關(guān)于磺胺類藥物殘留的檢測技術(shù)方法主要包括酶聯(lián)免疫法、高效液相色譜法、超高效液相色譜-串聯(lián)質(zhì)譜法(UPLC-MS/MS)等[10-16]。前兩種方法檢出限高,對復雜樣品的檢測靈敏度不高,難以適用基質(zhì)復雜的樣品;而UPLC-MS/MS能解決這一短板,適用于復雜組分中痕量藥物殘留測定。本研究采用分散固相萃取方法進行前處理,以不同配比的3種凈化填料(PSA,C18,MgSO4)為組合進行凈化效果對比試驗,優(yōu)化提取凈化條件,建立了一種UPLC-MS/MS測定水蛭中磺胺類藥物殘留的方法,能滿足水蛭中磺胺類藥物殘留的快速檢測需求。
1 儀器與試藥
1.1 儀器
1290超高效液相色譜-6460C三重四極桿串聯(lián)質(zhì)譜儀:配有電噴霧離子源(美國安捷倫);電子天平(梅特勒-托利多);移液器:20 μl,100 μl,1.0 ml(德國艾本德);渦旋混勻器(美國Scientific Industries);5810R離心機(德國艾本德);SBL-10DT超聲波清洗器(寧波新芝);IQ 7000 超純水機(美國默克密理博)。
1.2 材料
甲醇、乙腈、乙酸乙酯(色譜純,德國默克集團);甲酸、乙酸銨(色譜純,東京化成工業(yè));硫酸鈉、氯化鈉(分析純,國藥集團化學試劑公司);PSA,C18(月旭科技);MgSO4(分析純,天津百世)。
對照品:磺胺二甲嘧啶(批號:100411-200501,純度:100 %,中國食品藥品檢定研究院);磺胺間二甲氧嘧啶(批號:G122608,純度:99.5 %)、磺胺鄰二甲氧嘧啶(批號:G151421,純度:99.1 %)(Dr. Ehrenstorfer GmbH);磺胺嘧啶(批號:100026-201404,純度:99.7 %,中國食品藥品檢定研究院);磺胺間甲氧嘧啶(批號:97864,純度:99.1 %,曼哈格公司);磺胺喹噁啉(批號:G992252,純度:96.3 %)、磺胺甲噁唑(批號:G150000,純度:99.5 %)、磺胺甲基嘧啶(批號:G982705,純度:99.1 %)、、磺胺二甲異噁唑(批號:W1004574,純度:99.53 %)、磺胺噻唑(批號:G980077,純度:99.2 %)(Dr. Ehrenstorfer GmbH);實際檢測樣品購于市場。
2 方法與結(jié)果
2.1 色譜條件
色譜柱:ACQUITY BEH C18柱(1.7 μm,2.1 mm×100 mm);流動相A:含0.1 %甲酸-2 mmol/L乙酸銨-水,流動相B:含0.1 %甲酸-乙腈;梯度洗脫程序:0~2.0 min,15 %~30 %B;2.0~3.0 min,30 %~40 %B;3.0~4.5 min,40 %~90 %B;4.5~6.5 min,90 %B;6.5~7.0 min,90 %~15 %B;7.0~8.0 min,15 %B;流速:0.3 ml/min;柱溫:35 ℃;進樣體積:2 μl。
2.2 質(zhì)譜條件
離子源:噴射流電噴霧離子源(AJS ESI);離子極性:正離子;干燥氣流速:6 L/min;霧化器壓力:45 psi;離子源溫度:325 ℃;毛細管電壓:4000 V;鞘氣流速:10 L/min;鞘氣溫度:350 ℃;監(jiān)測模式:多反應(yīng)監(jiān)測模式(MRM);優(yōu)化后的質(zhì)譜參數(shù)見表1。

表1 10 種磺胺類藥物的質(zhì)譜參數(shù)
2.3 溶液制備
2.3.1 標準溶液制備 分別精密稱取各磺胺類藥物對照品適量,用乙腈配制成濃度約為 1 mg/ml的單標標準儲備液,置 4 ℃冰箱冷藏備用;取各單標標準儲備液適量,用乙腈配制成濃度約為 1 μg/ml的混合標準使用液,置 4 ℃冰箱冷藏備用;取混合標準使用液適量,用乙腈配制成濃度分別為0.05,2,5,10,20,30,40,50 ng/ml的標準曲線溶液。
2.3.2 供試品溶液制備 精確稱取1 g粉碎水蛭樣品置入50 ml離心管,加入3 ml Mcllvaine-Na2EDTA緩沖液,渦旋搖勻,使樣品充分潤濕復原,加入均質(zhì)子,渦旋1 min,精密加入10.00 ml乙腈,渦旋1 min,在800 W功率下超聲提取5 min ,加入1 g氯化鈉和4 g硫酸鈉,充分渦旋2 min,于4 ℃以3900 r/min離心10 min,取上清待凈化。
取待凈化液2 ml置入預先裝有100 mgPSA、150 mg C18、900 mg MgSO4的離心管,渦旋1 min,于4 ℃以3900 r/min離心10 min,上清過0.22 μm濾膜,供液相色譜串聯(lián)質(zhì)譜儀測定。
2.4 線性關(guān)系、檢出限及定量限
取混合標準使用液適量,配制成不同質(zhì)量濃度的系列標準曲線溶液,在優(yōu)化的儀器條件下測定,以各待測物質(zhì)譜響應(yīng)值為縱坐標(y),對應(yīng)濃度為橫坐標(x),繪制標準曲線(見表2)。由表2可見,10 種磺胺類藥物在0.05~50 ng/ml范圍內(nèi)線性關(guān)系良好,R2均大于0.99。以基質(zhì)標的3 倍和 10 倍信噪比時的檢測濃度為方法的檢出限和定量限,檢出限為0.19~1.16 μg/kg,定量限為0.56~3.95 μg/kg。

表2 10種磺胺類藥物的線性關(guān)系、檢出限及定量限
2.5 回收率與精密度
選用水蛭空白樣品作為基質(zhì)進行加標回收實驗,添加50,100,200 μg/kg 3個濃度水平的混合標準使用液,按照2.3.2項下方法與步驟處理加標樣品,上機測定,每個添加水平重復6次實驗,結(jié)果見表3。由表 3可見,10 種磺胺類藥物的平均回收率在65.2 %~98.0 %之間,日內(nèi)相對標準偏差(RSD)為1.9 %~8.3 %,日間相對標準偏差(RSD)為3.5 %~9.2 %,表明此法的準確度和精密度滿足定性定量檢測要求。
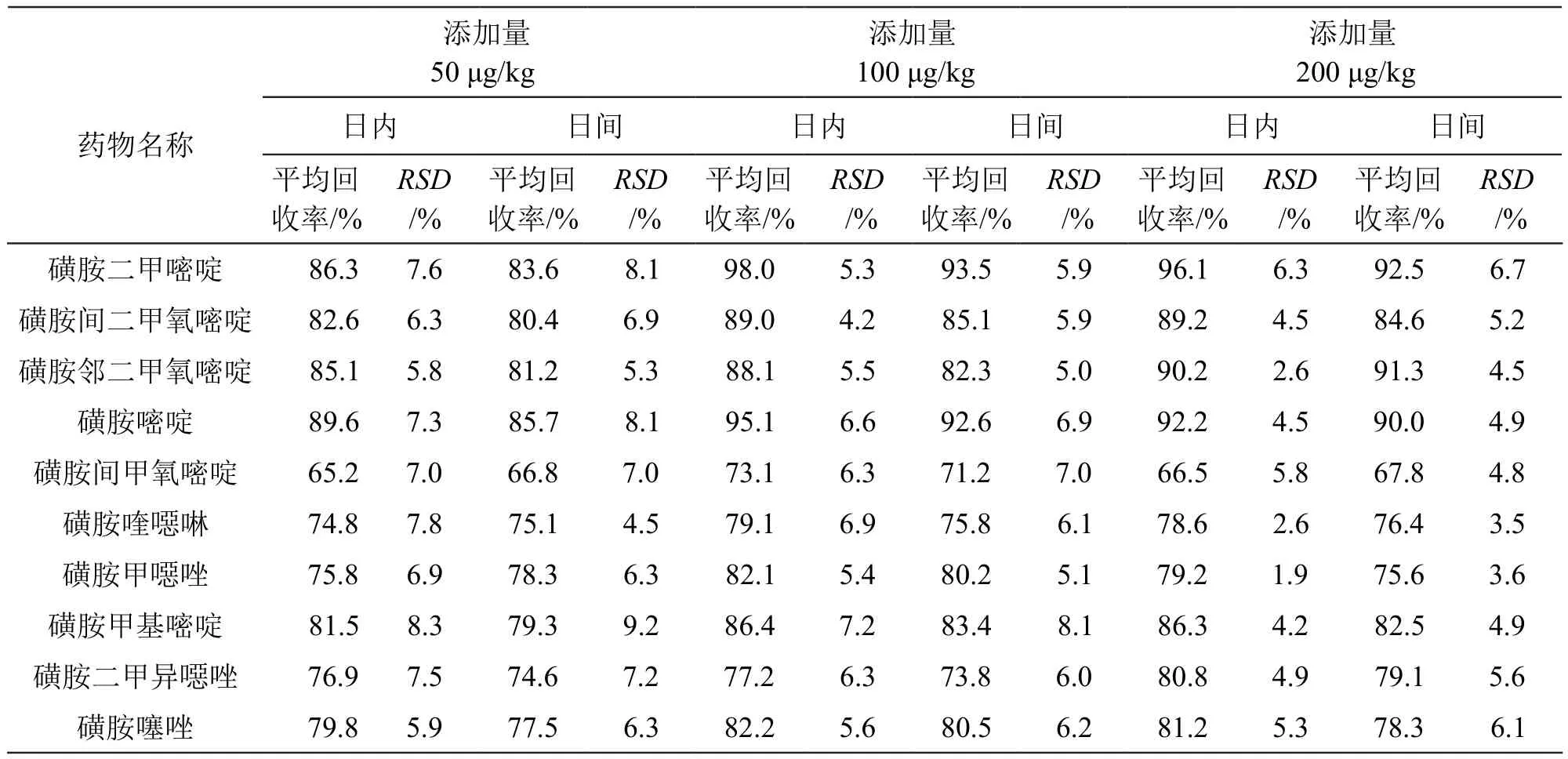
表3 10 種磺胺類藥物的回收率與精密度
2.6 耐用性
在質(zhì)譜條件不變情況下,分別測定用空白基質(zhì)溶液與乙腈稀釋配制的同濃度混合標準使用液,在流速、柱溫、進樣量等參數(shù)產(chǎn)生微小變動時,考察提取色譜圖、總離子流圖及質(zhì)譜響應(yīng)的變化。結(jié)果表明,這些參數(shù)發(fā)生微小變動對檢測結(jié)果的影響很小,表明此法有良好的耐用性。
2.7 磺胺類藥物殘留穩(wěn)定性
藥物在基質(zhì)殘留的穩(wěn)定性受樣品儲存條件的影響,磺胺類藥物本身化學性質(zhì)穩(wěn)定,在畜禽及水產(chǎn)品中的穩(wěn)定性已被研究證實。如2011年中國合格評定國家認可委員會組織了畜禽肉中磺胺類藥物殘留的穩(wěn)定性試驗的能力驗證工作,結(jié)果表明,磺胺類藥物在該類基質(zhì)中化學性質(zhì)穩(wěn)定[17]。有文獻采用水產(chǎn)品基質(zhì)加標的方法[18-19],考察磺胺類藥物殘留在常溫、反復凍融和低溫保藏等條件下的穩(wěn)定性,結(jié)果表明,磺胺類藥物殘留在水產(chǎn)品中的穩(wěn)定性較強,同理,樣品水蛭中磺胺類藥物殘留也比較穩(wěn)定。
2.8 樣品測定
采用建立的方法對市售10批次水蛭中10種磺胺類藥物進行測定,均未檢出上述10 種磺胺類藥物。
3 討論與結(jié)論
3.1 色譜質(zhì)譜條件優(yōu)化
3.1.1 色譜條件優(yōu)化 因磺胺類藥物有一定酸性,易溶于甲醇、乙腈等有機溶劑,因此,選擇了常用的反相 C18色譜柱進行分離。進一步考察不同流動相體系的分離效果和響應(yīng),在流動相的選擇上,為了提高目標化合物的離子化效率,對比了含不同濃度配比的甲酸和乙酸銨的水溶液,及含不同濃度甲酸的乙腈、甲醇對分離效果和質(zhì)譜響應(yīng)的影響。結(jié)果表明,有機相為0.1 %甲酸-乙腈,水相為0.1 %甲酸-2 mmol/L乙酸銨-水,各個被測藥物峰形和分離效果最佳,化合物的質(zhì)譜響應(yīng)值增強,因此,最終選擇0.1 %甲酸-2 mmol/L乙酸銨-水與含0.1 %甲酸-乙腈作為流動相。
3.1.2 質(zhì)譜條件優(yōu)化 通過對各藥物對照品溶液單標進樣,電噴霧正離子掃描模式下,優(yōu)化各個藥物的母離子、子離子、碎裂電壓、碰撞能量等質(zhì)譜參數(shù),并選擇干擾小且響應(yīng)值高的離子對作為定性離子和定量離子,建立多反應(yīng)監(jiān)測方法。
3.2 提取溶劑的選擇
比較乙腈、甲醇、乙酸乙酯3種常用提取溶劑的提取效率,結(jié)果表明,當甲醇為提取溶劑時,回收率為43 %~76 %,且大多數(shù)測定藥物的回收率小于70 %;當用乙酸乙酯提取時,回收率為49 %~79 %,且磺胺喹噁啉和磺胺甲噁唑的回收率均小于50 %;當乙腈為提取溶劑時,目標物的提取效果最好,回收率提高至79 %~87 %,原因可能是乙腈能很好地使樣品中蛋白質(zhì)變性,并在萃取鹽包及低溫高速離心的作用下,促使提取液的水相和有機相分層,進而通過離心去除蛋白脂肪等雜質(zhì),降低分析液中基質(zhì)干擾物的含量。因此最終選擇乙腈為提取溶劑(見圖1)。
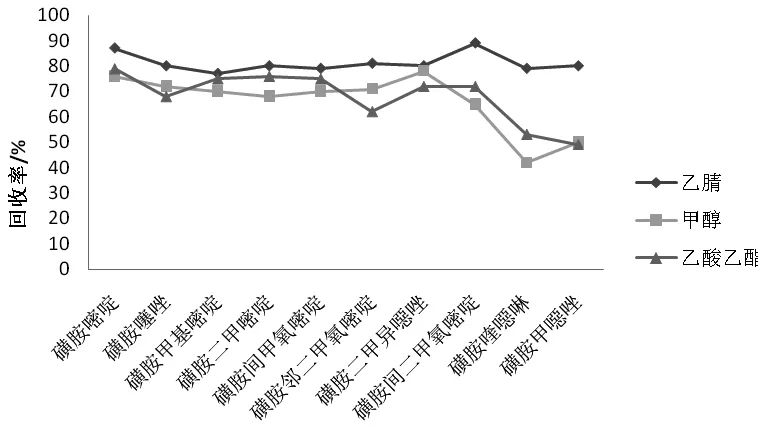
圖1 不同提取溶劑的提取效率
3.3 凈化方法的確定
設(shè)計9組不同配比的凈化劑組合(見表4),并考察了不同凈化劑組合的凈化效果,結(jié)果見圖2。用PSA 100 mg、C18150 mg、MgSO4900 mg組合凈化時,對目標檢測物凈化效果較好,10 種磺胺類藥物的回收率分布在80 %~100 %之間。其中,磺胺嘧啶和磺胺二甲嘧啶回收率接近100 %,最終選擇PSA 100 mg、C18150 mg、MgSO4900 mg組合作為凈化劑。
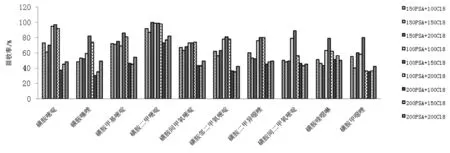
圖2 不同組合凈化劑提取回收率的對比

表4 不同組合的凈化劑
3.4 基質(zhì)效應(yīng)
水蛭作為基質(zhì)較復雜的樣品,進行質(zhì)譜分析時基質(zhì)殘留對目標物的離子化會產(chǎn)生影響,因此考察基質(zhì)對目標物檢測的影響。用空白基質(zhì)溶液與乙腈分別配制同濃度的標準溶液,以兩者質(zhì)譜響應(yīng)值比值的百分數(shù)評價基質(zhì)效應(yīng)(ME),若ME大于100 %,則有基質(zhì)增強效應(yīng),相反,則為抑制效應(yīng);若ME接近100 %,則不存在基質(zhì)效應(yīng)。本實驗測試了10 ng/ml和20 ng/ml兩個濃度水平,結(jié)果表明,20 ng/ml時,除磺胺二甲嘧啶有基質(zhì)效應(yīng)增強效應(yīng)外,其余目標待測物均有不同程度弱的基質(zhì)抑制效應(yīng),其ME大于75 %。10 ng/ml時,磺胺二甲嘧啶和磺胺嘧啶ME接近100 %,無基質(zhì)效應(yīng);磺胺間甲氧嘧啶、磺胺喹噁啉、磺胺甲噁唑、磺胺二甲異噁唑ME在70 %~80 % ,表明有中等基質(zhì)抑制效應(yīng),其余目標待測物ME為80 %~89 %,有弱的基質(zhì)抑制效應(yīng)。雖然各待測物有不同程度弱或中等基質(zhì)效應(yīng),但均在可接受的范圍內(nèi),對檢測結(jié)果不產(chǎn)生影響(見圖3)。
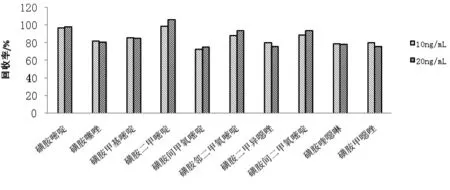
圖3 10種磺胺類藥物的基質(zhì)效應(yīng)
3.5 結(jié)論
綜上,本研究首次建立了一種基于分散固相萃取凈化的超高效液相色譜-串聯(lián)質(zhì)譜法測定水蛭中10種磺胺類藥物殘留的分析方法。該分析方法以緩沖鹽溶液復原干燥樣品,以乙腈為提取溶劑變性沉淀樣品中蛋白質(zhì),通過加入鹽包,使提取液中的水相和有機相在飽和鹽存在下實現(xiàn)分層,通過離心去除蛋白質(zhì)。以分散固相萃取為凈化手段,選擇100 mg PSA、150 mg C18、900 mg MgSO4組合為凈化劑,對提取液進行進一步凈化。對建立的方法進行方法學驗證,其各項參數(shù)均滿足準確定量檢測的要求。本研究建立的方法操作簡便、準確性良好,可為中藥材水蛭中磺胺類藥物殘留的快速檢測提供技術(shù)支撐。
