計算機虛擬篩選技術在中藥毒性研究中的探索與思考
2023-06-19 02:49:00錢文秀閻星旭張文青賈國香宋麗麗李遇伯
中草藥 2023年12期
錢文秀,閻星旭,張文青,賈國香,趙 珊,宋麗麗,李遇伯
計算機虛擬篩選技術在中藥毒性研究中的探索與思考
錢文秀,閻星旭,張文青,賈國香,趙 珊,宋麗麗*,李遇伯*
天津中醫藥大學中藥學院,天津 301617
近年來,中藥安全性問題逐漸成為社會關注的熱點,尤其是中藥毒性研究更是中藥研究的重點。因此,尋找高效的技術輔助中藥毒性研究尤為關鍵。計算機虛擬篩選技術具有高效、便捷等優點,經過多年的發展,其技術理論已經趨于成熟,且在多個領域已被成功應用。通過對分子對接、機器學習、毒效團和分子相似性方法4種主要的計算機虛擬篩選技術進行綜述,并分析它們在中藥毒性研究領域的應用和前景,為從事中藥毒性研究的科研人員提供參考。
虛擬篩選;分子對接;機器學習;毒效團;分子相似性;中藥毒性研究
中藥作為我國的寶貴財富,在治療某些慢性或特殊性疾病方面具有顯著效果,但中藥體系復雜,其在發揮強大治療作用的同時,安全性問題也不容忽視[1]。近年來,科研人員越來越重視中藥毒性問題的研究,毒性研究方向也逐漸趨于多元化,且已經取得了顯著成果[2],但仍然存在一些問題:(1)中藥毒性成分結構復雜多樣,并且多數中藥毒性成分未知[3];(2)多數中藥毒性機制不明,而且因中藥多靶點、多途徑、多效應、多環節的作用方式致使毒性機制復雜多樣[4-5],因而造成毒性機制研究過程較復雜、困難;(3)以往被認定為無毒的中藥也被報道出能夠產生不良反應等問題[6]。因此,探索新的技術來輔助中藥毒性研究是一項極其緊迫且重要的工作。
計算機虛擬篩選技術融合了化學、分子生物學、毒理學、統計學及計算機科學等多個學科,通過構建數學及計算機模型來實現對毒性化合物、毒副作用及毒性機制初步探究的目的。目前,該技術已在化學藥毒理學研究、農藥風險評估、食品毒理學、環境管理等多個領域被廣泛應用[7-13]。根據建模原理的不同,虛擬篩選技術主要分為2類(圖1),第1種為基于受體的大分子虛擬篩選技術,也被稱為基于結構的虛擬篩選技術,是目前中藥研究中應用較為廣泛的一類篩選技術,使用該類技術進行毒性成分篩選時需準備毒性機制清晰的受體生物大分子,這類技術包括分子對接[14-15];第2種為基于配體的小分子虛擬篩選技術,該類篩選技術主要是通過分析化學成分間的相似性程度實現毒性成分篩選,進而達到研究成分結構特點與毒性作用間關系的目的,這類篩選技術主要包括機器學習、毒效團、分子相似性方法等[14]。本文擬對目前4種主要的虛擬篩選技術及其在中藥毒性研究中的具體應用情況進行綜述,并對虛擬篩選技術在中藥毒性研究中的應用前景進行分析,為今后從事相關研究的研究者提供思路。
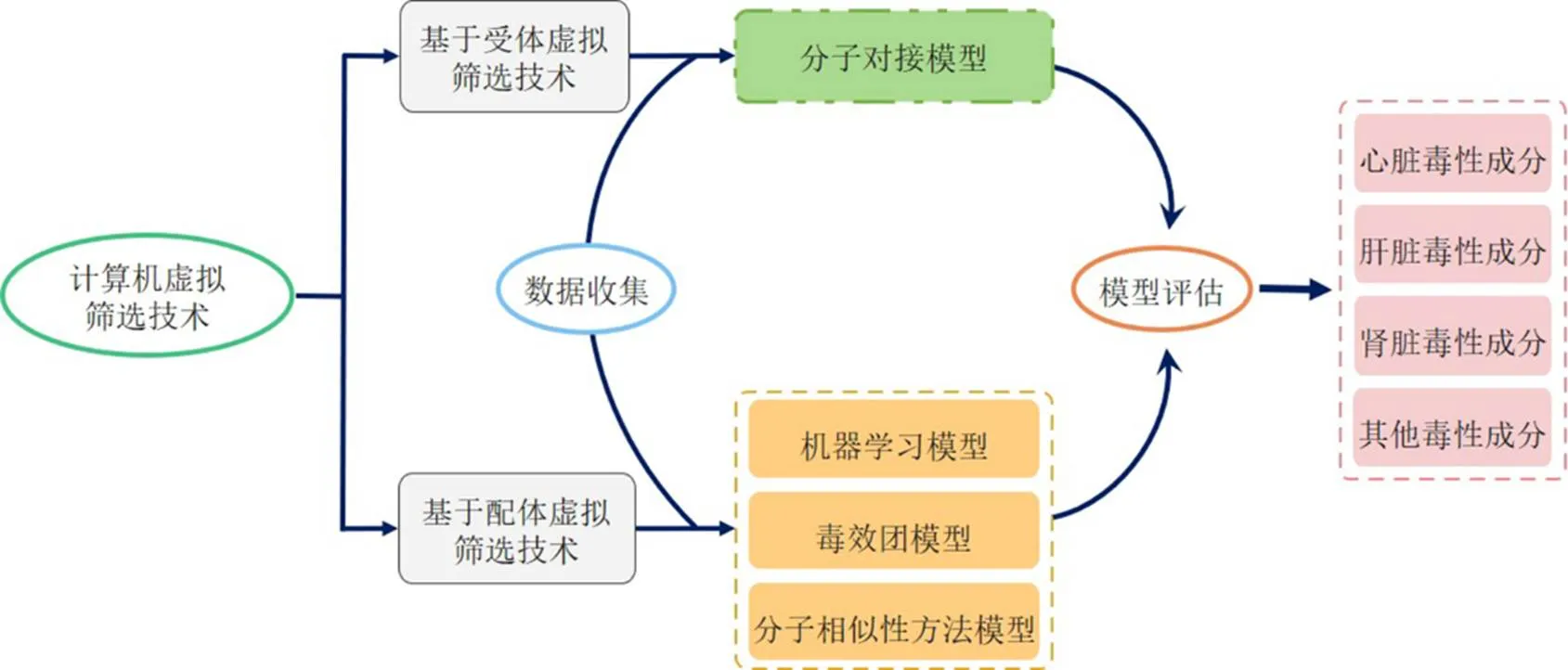
圖1 計算機虛擬篩選技術概況
1 分子對接
分子對接的工作原理是依據給定的靶標篩選出與之匹配的潛在化學成分,該技術可用于模擬大分子受體與小分子配體間的作用方式,發現目標成分,指導成分結構優化,對活性和非活性成分進行分類(如篩選出毒性成分)及藥物生物活性機制(毒性機制)等研究[16-20]。分子對接作為較早被應用于藥物研究的虛擬篩選技術,經過幾十年的發展,其技術理論已較為成熟和完善。如今,分子對接技術以其高效、快速、便捷等優勢吸引了中藥領域科研人員的關注,在中藥毒性研究中占據重要位置。
分子對接的操作步驟可大致分為2個階段[21],(1)通過構象搜索獲得可能的受體及配體的結合模式;(2)利用打分函數對產生的構象進行打分,現有的打分函數大致分為基于力場、知識、經驗及機器學習4種,選擇恰當的打分函數是保證選擇最佳構象結合模式的關鍵所在[22]。分子對接作為一種高效的科研輔助工具,將其運用于中藥毒性研究時應當在符合中藥特性的前提下,充分考慮實驗各階段可能涉及的問題,包括對接前的數據準備與處理,對接時的參數選擇及對接后的模型驗證和評估等工作,以期能夠盡可能的降低實驗誤差,進而保證分子對接模型是有意義的,結果是準確可靠的。
1.1 心臟毒性成分的篩選
石洲[23]通過分子對接技術聯合結構毒-效關系對川烏的毒性成分進行篩選分析,具體為應用Autodock Vina軟件的半柔性對接方式,采用快速梯度優化算法,將對接次數及exhaustiveness值分別調整為20和10,結果篩選出烏頭堿、中烏頭堿、次烏頭堿和去甲烏藥堿等心臟毒性成分,然后通過分析分子對接的活性基團位置,明確C-19雙酯型二萜類生物堿是川烏造成心臟毒性貢獻最大的成分。表明分子對接技術不僅可以篩選出中藥潛在的心臟毒性成分,還可以用于確定中藥毒性成分的結構特點。
1.2 肝毒性成分的篩選
汪祺等[24-26]選擇性收集了文獻及數據庫中的48種何首烏成分,然后以多藥耐藥相關蛋白2/多藥耐藥相關蛋白3、有機陰離子轉運多肽1B1/有機陰離子轉運多肽1B3和法尼醇X受體作為靶標,借助Discovery Studio 2.5軟件分別進行3次分子對接,根據打分值的排序結果篩選出打分值較高且在何首烏中含量較高的成分,初步確定何首烏中存在肝毒性風險的成分,隨后采用體外肝細胞毒性實驗對初步篩選出的潛在肝毒性成分進行研究以驗證虛擬篩選結果的準確性。結果證實,細胞實驗與分子對接結果基本一致,進一步說明分子對接技術用于篩選中藥肝毒性成分的可靠性。
Liu等[27]使用分子對接技術對采用超高效液相色譜-四級桿飛行時間質譜鑒定出的14種八角楓化學成分進行肝毒性成分篩選,借助AutoDock Tool 1.5.6軟件和Lamarck遺傳算法構建模型進行分子對接計算,考察最佳的結合位點,結果發現鬼臼毒素-4---葡萄糖苷、西藏鬼臼脂醇、3,4′-,-二去甲基鬼臼毒素、鬼臼脂毒酮及鬼臼毒素等成分能夠與肝毒性靶點有較好的結合,因此初步確定上述成分為八角楓的肝毒性成分。
1.3 其他毒性成分的篩選
分子對接除可以單獨使用外,還常與其他方法聯合應用,使得篩選效率及準確率進一步提高,較為常見的組合方式是與網絡藥理學(網絡毒理學)的聯合應用,首先借助網絡藥理學(網絡毒理學)篩選核心毒性靶標,并通過構建“活性成分-毒性靶標-通路”網絡了解成分與靶標間的關系;隨后基于得到的核心毒性靶標和活性成分,采用分子對接技術進行毒性成分篩選[28-30]。張丹等[31]為研究昆明山海棠的毒性機制,運用分子對接和網絡藥理學對昆明山海棠的毒性成分進行篩選研究,發現雷公藤堿乙、雷酚內酯和山海棠二萜內酯A能夠與細胞周期蛋白D1、促分裂原活化蛋白激酶8、血管內皮生長因子A、腫瘤蛋白p53、白細胞介素-6、腫瘤壞死因子等毒性靶標有著較好的相互作用與結合活性,從而初步確定以上3種成分可能為昆明山海棠的毒性成分。上述實驗表明分子對接作為一種技術理論較成熟的虛擬篩選技術,既可以篩選出毒性表現明確的中藥成分,又可以成功篩選出毒性表現不明的中藥成分。
2 機器學習
機器學習的基本原理是提取化合物的結構及理化性質特征,然后通過總結配體的特征選用合適的算法構建模型,最后利用模型對化合物進行篩選。目前機器學習的學習方法主要有監督學習、半監督學習、無監督學習及強化學習等[32-35]。不同的學習方法有不同的算法,如監督學習常用算法包括決策樹、支持向量機(support vector machine,SVM)、線性回歸、樸素貝葉斯;無監督學習常用算法有K-最鄰近方法和聚類算法[34,36]。因該技術能夠通過學習對模型進行不斷優化,因此篩選結果的準確率較高[37]。
隨著機器學習技術的快速發展,在此基礎上出現了一個新的分支技術——深度學習。深度學習是基于人工神經網絡(artificial neural network,ANN)算法來構建模型,該技術有監督、無監督或二者兼備的學習方法,對于處理大體量且復雜的毒性數據集具有顯著優勢,且研究結果的準確率也大幅度提高。鑒于深度學習的以上優點將其用于中藥毒性成分篩選研究具有廣闊的前景[34,38]。
定量構效關系(quantitative structure activity relationship,QSAR)利用統計學方法和機器學習算法構建模型,分析化合物的特征與結構參數,從而篩選具有相似理化性質及生物活性的化合物,該技術還可以解釋化合物的結構變化與性質變化間的關系[39]。QSAR有助于篩選毒性特征相似的中藥毒性化合物,指導毒性化合物的結構優化。同時,由于QSAR是基于配體實現的虛擬篩選技術,所以在應用中無需考慮靶蛋白結構等問題。另外,當應用環境強調化合物結構與毒性間的關系時QSAR也可定義為定量毒性結構關系[40]。經過幾十年的發展,QSAR技術經過不斷地完善和升級,現在QSAR模型已經從2D-QSAR、3D-QSAR發展到了4D-QSAR、5D-QSAR、6D-QSAR等[41-45]。QSAR模型在化學、生物、醫藥、農業、環境等領域有著廣泛地應用,并且在中藥毒性研究中也發揮著越來越重要的作用。
2.1 心臟毒性成分的篩選
馬會利[46]從藥物副作用數據庫、OFFSIDES、比較毒理學數據庫中收集到1555個心臟毒性化合物,RADER數據庫中收集到4670個非活性化合物,設置心臟毒性成分與非毒性成分數量3∶1的比例構建各數據集,以4665個化合物構建訓練集,1560個化合物構建測試集,然后使用MOE 2009和PaDEL-Descriptor 2.21軟件計算分子描述符,借助Orange Canvas 2.7版的邏輯回歸、隨機森林和K-最近鄰3種算法構建了6個機器學習分類模型,另外采用5倍交叉驗證與測試集驗證這2種方法進行模型驗證,篩選出PaDEL_LFS_LR+MACCS與PaDEL_LFS_LR+SubFP 2個最優模型對從巴豆、斑蝥、蓖麻子、蒼耳子等20味中藥中收集的708個成分進行心臟毒性成分篩選,最終篩選出亞油酸、棕櫚酸、硬脂酸、β-谷固醇、谷固醇、油酸6個潛在心臟毒性成分。馮小龍[47]對從《中國藥典》2015年版中收集到的45種含揮發油類中藥的181個主要化學成分進行毒性成分篩選,使用ADMET Predictor 9.5軟件構建基于ANN算法的QSAR模型,選用人類快速延遲整流性鉀通道基因作為心臟毒性評價的標準,篩選出木蘭脂素、榿木酮、去氫二異丁香酚等6個化學成分可能具有心臟毒性。這些研究案例表明QSAR模型可以實現從較大規模的化學物集合中篩選出中藥毒性成分,為中藥毒理學研究提供了一個高效且較為可靠的技術。
2.2 肝毒性成分的篩選
為了探索中藥-化學藥組合化合物數據集的肝毒性成分篩選方案,Chen等[48]從數據庫和文獻中收集了1505個化合物作為訓練集,530個化合物作為測試集,2個數據集中肝毒性與非肝毒性成分的數量比值分別為1125∶400和380∶130,然后分別采用隨機梯度下降、K-最近鄰、SVM、樸素貝葉斯、決策樹、隨機森林、ANN、自適應提升、邏輯回歸等算法構建機器學習和深度學習模型,該研究證明將機器學習模型用于篩選肝毒性成分有廣闊的應用前景,而且通過對比分析發現深度學習模型的性能更優。
Zhao等[49]收集含有127種肝毒性成分的陽性數據集,其中含有11種肝毒性中藥成分,及含有65種與肝毒性無關的陰性成分數據集,使用Mold軟件計算分子描述符并挑選出98個最佳的分子描述符用于構建基于隨機森林算法的QSAR模型,最后通過外部實驗進行模型驗證,結果發現QSAR模型篩選出天然肝毒素數據集中的14種肝毒性成分。馮小龍[47]使用ADMET Predictor 9.5軟件構建了基于ANN算法構建QSAR模型,以丙氨酸氨基轉移酶、谷氨酸轉移酶和乳酸脫氫酶同時升高作為肝毒性評價指標,從181個中藥成分中篩選出63個肝毒性成分。機器學習可以通過學習和選擇合適的算法來不斷優化模型,因此采用該技術構建模型篩選中藥肝毒性成分時結果的準確率較高。
2.3 腎毒性成分的篩選
構建QSAR模型用以中藥毒性研究時若實驗數據不足或是采用化學藥物構建模型則可能會造成偏差的產生,Sun等[50]為了驗證這一問題,分別用天然產物(128種)、藥物(484種)及二者混合這3類數據構建訓練集,3個數據集分別使用ANN和SVM 2種機器學習算法構建QSAR模型考察其對中藥腎毒性成分的篩選能力,結果發現天然產物模型的準確性較好,說明QSAR模型可較好地適用于中藥腎毒性成分的篩選。Yang等[51]首先使用ADMET Predictor軟件構建QSAR模型對新分離的26種決明子成分進行腎毒性成分初步篩選,隨后將篩選出的潛在毒性成分與人腎小管上皮HK-2細胞培養對篩選結果進行驗證,結果確定了10種具有潛在腎毒性的成分。中藥成分與化學藥成分的結構特點有所區別,因此使用機器學習技術進行中藥毒性成分篩選時,應當采用較多的中藥成分構建模型以保證研究結果的準確性。
2.4 其他毒性成分的篩選
眾所周知馬兜鈴酸是一種強致腎毒性和致癌的成分。Xu等[52]為篩選出馬兜鈴酸類似物,借助Python 3.7中的RDKit庫提取出馬兜鈴酸類成分的3個最大公共子結構,進而篩選出存在于44種中藥中的238種馬兜鈴酸類成分,提示這些被篩選出的類似物可能具有和馬兜鈴酸相同的毒性作用。機器學習技術不僅能夠實現毒性成分的篩選,還有助于毒性成分后續的毒性作用機制研究。
3 其他虛擬篩選技術
除以上介紹的較為常用的虛擬篩選技術外,還有毒效團和分子相似性方法這2種技術。雖然還未被廣泛地應用于中藥毒性研究,但是通過分析這2種技術在其他領域的應用情況發現,有望將這些技術應用于中藥毒性成分的篩選,且同樣具有高效、便捷的特點,值得中藥科研人員的探索及使用[53-56]。
3.1 毒效團
毒效團是從藥效團技術衍生出來的基于配體的虛擬篩選技術,其基本原理與藥效團相似。它是根據化合物中發揮毒性作用或經過代謝后發揮毒性作用的基團或亞結構為基礎構建模型,篩選出含有這些結構的潛在毒性化合物,然后研究化合物關鍵結構與生物活性(毒性)間的關系,進而揭示化合物的毒性機制[57]。另外,這些發揮毒性作用的基團或亞結構也被稱為警示結構、毒性片段、毒性基團,是藥物產生毒副作用的主要原因之一[57-59]。
雖然,迄今為止毒效團技術用于中藥毒性研究較少,但是通過考察毒效團模型在其他領域的成功應用案例,確定將該模型用于中藥毒性研究是切實可行的,其基本工作流程可大致分為以下4個步驟[53-54],(1)數據收集:根據化合物的毒性效應強弱劃分訓練集及測試集,收集以上化合物的2D結構,確定模型構建需要的化合物活性值(半數致死量、半抑制濃度等);(2)構建模型:確定建模軟件,設置參數構建模型,選擇用于構建模型的特征元素(氫鍵供體、氫鍵受體、芳香環、疏水基團、正電荷基團、負電荷基團)且最多可選擇5個特征元素;(3)模型驗證及評價:挑選最優模型;(4)篩選活性成分:使用最優模型對預測集成分進行匹配,篩選出與模型匹配最佳的活性化合物。
毒效團的出現為后續的實驗發展方向提供了清晰的指引,研究者可通過分析被篩選出的含有警示結構的毒性成分特性進行后續實驗內容設計。總體來說,毒效團可為后續實驗提供2種研究思路,首先是對于含有警示結構但既可起到治療作用又可產生不良反應的成分可采用結構優化的方式以降低其毒性;其次是對于含有警示結構且只產生毒副作用的成分則采取消除該成分的操作。
3.2 分子相似性
分子相似性方法依據化合物結構特點的相似性程度篩選出具有類似理化性質和生物活性的化合物,該類技術對結構特征匹配的精確性具有較高的要求[32]。分子相似性方法具體分為結構相似性搜索和活性片段搜索2種類型[60-62]。以上2種類型相似性方法的建模流程是相同的[55],首先需要針對某一種活性來篩選化合物以構建數據集,然后選擇用于建模的分子結構參數,如分子形狀指數、電拓撲指數等,最后建模并計算相似性參數。目前常用的可以實現該技術的平臺有MolPrint 2D、ChemMappe、eSHAFTS、Me2Explorer等[63-66]。
4 虛擬篩選技術應用分析
從文獻檢索的過程中發現,虛擬篩選技術因其便捷、快速、高效、準確率較高等特點,已被成功運用于心臟、肝臟、腎臟毒性等類型的中藥毒性成分篩選,其中將該技術用于中藥肝毒性成分篩選的研究課題居多,這可能與造成肝毒性的中藥較多有關[5,67]。因為用于建模的肝毒性基礎毒理學實驗數據較多,所以使用虛擬篩選技術進行肝毒性成分篩選時的可行性及準確性更高。眾所周知,中藥毒性表現復雜多樣,且一種有毒中藥的毒性表現往往不止一種,因此盡可能全面的了解中藥毒性,才能保證中藥的正確使用。參考虛擬篩選技術在中藥肝毒性成分篩選中的成功案例,將虛擬篩選技術用于更多種毒性類型的成分篩選研究,如生殖毒性、神經毒性、致癌性等,這是未來虛擬篩選技術的一個應用方向。
虛擬篩選技術在中藥毒性領域中的應用仍處于起步階段,采用虛擬篩選技術進行中藥毒性成分篩選的案例是較少的,而且本文中介紹的毒效團和分子相似性方法還未被廣泛應用于中藥毒性研究。因此,研究者可以在充分考慮每種虛擬篩選技術的優點與不足(表1)的基礎上,結合分析中藥的特點,同時參考這些技術在其他領域的成功運用案例[53-54,68],開發出適用于中藥特性的毒性研究方案,這將是中藥研究者關注的一個重要研究方向,也是虛擬篩選技術發展的必然趨勢。

表1 計算機虛擬篩選技術特點
5 結語與展望
中藥毒性研究既是促進中藥新藥研發與現代化發展的基礎,也是推動中藥研究的重點及難點所在[69]。計算機虛擬篩選技術恰當地將多學科融合在一起,極大地減少了使用傳統毒性實驗方法帶來的工作量大、實驗周期長、工作效率低及資源浪費等問題。目前,虛擬篩選技術在多個領域發揮著不可忽視的重要作用,尤其是近年來,該技術在中藥毒性研究領域的應用極大地提高了工作效率。但是,需要明確的問題是這項技術的出現并不是為了取代傳統的毒性研究方法,而是為了借助其強大的輔助作用來促進中藥毒性研究的快速發展。
本文介紹了分子對接、機器學習、毒效團和分子相似性方法4種主要的計算機虛擬篩選技術,并列舉了在中藥毒性研究中的成功案例,為虛擬篩選技術能夠高效地應用于中藥毒性研究提供參考思路。隨著未來計算機虛擬篩選技術理論的不斷優化和升級,將會越來越適應中藥毒性研究的特點,成為未來中藥快速發展進程中不可或缺的重要手段。
利益沖突 所有作者均聲明不存在利益沖突
[1] 張冰, 呂錦濤, 張曉朦, 等. 基于“藥性”的中藥“毒-效”認知與藥物警戒思考 [J]. 中國藥物警戒, 2021, 18(5): 411-415.
[2] 韓嵐, 林朝展, 趙海霞, 等. 2012—2021年國家自然科學基金中藥毒理學研究方向資助項目分析 [J]. 中國中藥雜志, 2022, 47(5): 1415-1420.
[3] 孟驛佳, 康樂, 王媛, 等. 2020年版《中國藥典》(一部)有毒中藥毒性成分及毒性特點分析 [J]. 中藥藥理與臨床, 2023, 39(1): 99-104.
[4] 何月, 許娜, 吳德歡, 等. 神經毒性中藥中毒機制與減毒策略的研究進展 [J]. 中南藥學, 2017, 15(7): 940-942.
[5] Pan X Q, Zhou J, Chen Y,. Classification, hepatotoxic mechanisms, and targets of the risk ingredients in traditional Chinese medicine-induced liver injury [J]., 2020, 323: 48-56.
[6] 柏兆方, 孟雅坤, 賀蘭芝, 等. 傳統無毒中藥誘導的免疫特異質型肝損傷及其機制假說 [J]. 中國藥學雜志, 2017, 52(13): 1105-1109.
[7] 姜允申. 計算毒理學和量子毒理學在我國環境醫學與藥物學研究中的應用及前景 [A] // 中國環境科學學會. 2020中國環境科學學會科學技術年會論文集 (第4卷) [C]. 南京: 中國環境科學學會, 2020: 85-89.
[8] 孫亞奇, 陸啟榮, 劉愛梅, 等. 藥物代謝誘導毒性的體外預測方法研究進展 [J]. 中國藥理學與毒理學雜志, 2019, 33(11): 1000-1006.
[9] 于洋, 左平春, 張楠, 等. 計算毒理學在農藥風險評估中的應用 [J]. 農藥科學與管理, 2017, 38(4): 24-30.
[10] Silva M H. Use of computational toxicology (CompTox) tools to predicttoxicity for risk assessment [J]., 2020, 116: 104724.
[11] Vuorinen A, Bellion P, Beilstein P. Applicability ofgenotoxicity models on food and feed ingredients [J]., 2017, 90: 277-288.
[12] Forest V. Experimental and computational nanotoxicology- complementary approaches for nanomaterial hazard assessment [J]., 2022, 12(8): 1346.
[13] 鄭玉婷, 王寶成, 于洋, 等. 一種篩選具有潛在持久性、遷移性和毒性(PMT) 新污染物的計算毒理學模型工具 [J]. 生態毒理學報, 2022, 17(3): 111-120.
[14] De León G, Fr?hlich E, Salar-Behzadi S. Bitter taste: A review on virtual ligand screening and characterization methods for TAS2R-bitterant interactions [J]., 2021, 600: 120486.
[15] Maia E H B, Assis L C, de Oliveira T A,. Structure-based virtual screening: From classical to artificial intelligence [J]., 2020, 8: 343.
[16] 周玉玳, 戴雙雄, 佟斌, 等. 光譜學與分子對接結合解釋生物大分子與配體間作用機制的研究進展 [J]. 影像科學與光化學, 2021, 39(3): 337-347.
[17] 朱銳靈, 沈悅, 馬飛鴻, 等. 分子對接技術在中藥抗炎活性成分篩選和作用機制研究中的應用 [J]. 中國藥理學與毒理學雜志, 2018, 32(6): 497-506.
[18] 劉景陶, 柳耀花. 通過分子對接確定并優化先導化合物 [J]. 科技創新與應用, 2018(3): 3-4.
[19] 汪祺. 基于UGT1A1抑制作用及體內外代謝產物探索何首烏肝毒性機制及毒效物質 [D]. 北京: 北京中醫藥大學, 2018.
[20] 孫雨晴, 張鑫, 沈曉帆, 等. 體外實驗和計算模擬研究七葉皂苷的腎細胞毒性機制 [A] // 2021 (第五屆) 毒性測試替代方法與轉化毒理學 (國際) 學術研討會會議論文集 [C]. 沈陽: 中國毒理學會, 2021: 112-113.
[21] 許先進, 王存新. 分子對接方法在藥物發現之外領域的應用 [J]. 北京工業大學學報, 2017, 43(12): 1872-1880.
[22] Li J, Fu A L, Zhang L. An overview of scoring functions used for protein-ligand interactions in molecular docking [J]., 2019, 11(2): 320-328.
[23] 石洲. 基于網絡藥理學和分子對接的川烏毒效成分及機制研究 [D]. 成都: 電子科技大學, 2021.
[24] 汪祺, 文海若, 馬雙成. 以MRP2/MRP3轉運體為作用靶點的何首烏中肝毒性成分篩選 [J]. 中國現代中藥, 2022, 24(4): 622-628.
[25] 汪祺, 文海若, 馬雙成. 以OATP1B1/OATP1B3轉運體為作用靶點的何首烏肝毒性成分篩選 [J]. 藥物評價研究, 2022, 45(2): 227-233.
[26] 汪祺, 文海若, 馬雙成. 以法尼醇X受體為作用靶點的何首烏中肝毒性成分的篩選 [J]. 中國藥物警戒, 2022, 19(6): 630-634.
[27] Liu C X, Zhang C N, He T,. Study on potential toxic material base and mechanisms of hepatotoxicity induced bybased on toxicological evidence chain (TEC) concept [J]., 2020, 190: 110073.
[28] Zhang X Y, Wang S, Shu L X,. Rapid screening of hepatotoxic components inbased on “component-target-pathway” network [J]., 2022, 219: 114968.
[29] 牛明, 張斯琴, 張博, 等.《網絡藥理學評價方法指南》解讀 [J]. 中草藥, 2021, 52(14): 4119-4129.
[30] 曹站霞, 陳毓龍, 李偉霞, 等. 基于網絡藥理學和細胞試驗探討補骨脂致肝損傷的毒理機制 [J]. 中國現代應用藥學, 2022, 39(16): 2052-2062.
[31] 張丹, 董一珠, 呂錦濤, 等. 基于網絡藥理學與分子對接方法探討昆明山海棠的毒性機制 [J]. 北京中醫藥大學學報, 2019, 42(12): 1006-1015.
[32] Yang X, Wang Y F, Byrne R,. Concepts of artificial intelligence for computer-assisted drug discovery [J]., 2019, 119(18): 10520-10594.
[33] 周玥, 張心苑, 毛雪石. 機器學習在創新藥物研發中的應用進展 [J]. 醫學信息學雜志, 2020, 41(8): 25-28.
[34] Vo A H, van Vleet T R, Gupta R R,. An overview of machine learning and big data for drug toxicity evaluation [J]., 2020, 33(1): 20-37.
[35] Choi R Y, Coyner A S, Kalpathy-Cramer J,. Introduction to machine learning, neural networks, and deep learning [J]., 2020, 9(2): 14.
[36] Idakwo G, Luttrell J, Chen M J,. A review on machine learning methods fortoxicity prediction [J]., 2018, 36(4): 169-191.
[37] 高宇, 王鳳雪, 劉海波. 虛擬篩選技術在天然產物新藥研發中的應用 [J]. 國際藥學研究雜志, 2020, 47(8): 602-608.
[38] 顏彩琴, 范睿琦, 寧雨坪, 等. 深度學習模型在中藥毒性預警中的應用和前景 [J]. 中國藥理學與毒理學雜志, 2022, 36(3): 231-240.
[39] Gini G. QSAR methods [J]., 2022, 2425: 1-26.
[40] Deeb O, Goodarzi M.quantitative structure toxicity relationship of chemical compounds: Some case studies [J]., 2012, 7(4): 289-297.
[41] 施海楓, 劉永香, 涂文通. 國內有關黃酮類化合物及其衍生物的2D-QSAR和3D-QSAR研究進展 [J]. 廣東化工, 2013, 40(24): 70-71.
[42] Sato A, Miyao T, Jasial S,. Comparing predictive ability of QSAR/QSPR models using 2D and 3D molecular representations [J]., 2021, 35(2): 179-193.
[43] Fourches D, Ash J. 4D-quantitative structure-activity relationship modeling: Making a comeback [J]., 2019, 14(12): 1227-1235.
[44] Bak A. Two decades of 4D-QSAR: A dying art or staging a comeback? [J]., 2021, 22(10): 5212.
[45] 李仁利. 三維以上定量構效關系 [J]. 國際藥學研究雜志, 2007, 34(4): 241-245.
[46] 馬會利. 化合物心臟毒性虛擬預測模型的構建及其應用研究 [D]. 廣州: 廣州中醫藥大學, 2018.
[47] 馮小龍. 揮發油類中藥主要化學成分安全性預測研究 [J]. 世界科學技術—中醫藥現代化, 2020, 22(9): 3065-3072.
[48] Chen Z, Zhao M Z, You L Z,. Developing an artificial intelligence method for screening hepatotoxic compounds in traditional Chinese medicine and Western medicine combination [J]., 2022, 17(1): 58.
[49] Zhao P, Liu B, Wang C Y. Hepatotoxicity evaluation of traditional Chinese medicines using a computational molecular model [J]., 2017, 55(9): 996-1000.
[50] Sun Y, Shi S, Li Y,. Development of quantitative structure-activity relationship models to predict potential nephrotoxic ingredients in traditional Chinese medicines [J].2019, 128: 163-170.
[51] Yang J L, Wang S, Zhang T,. Predicting the potential toxicity of 26 components inusingandapproaches [J]., 2021, 2: 237-245.
[52] Xu T J, Chen W M, Zhou J H,. Computational analysis of naturally occurring aristolochic acid analogues and their biological sources [J]., 2021, 11(9): 1344.
[53] Pramanik S, Roy K. Exploring QSTR modeling and toxicophore mapping for identification of important molecular features contributing to the chemical toxicity in[J]., 2014, 28(2): 265-272.
[54] Kar S, Roy K. First report on predictive chemometric modeling, 3D-toxicophore mapping andscreening ofbasal cytotoxicity of diverse organic chemicals [J]., 2013, 27(2): 597-608.
[55] 喬連生, 郭亦然, 張燕玲. 基于片段搜索和相似性搜索的抗哮喘中藥發現研究 [J]. 中國實驗方劑學雜志, 2013, 19(12): 310-314.
[56] 孔德毅. 分子相似性網絡中關鍵化合物發現算法研究 [D]. 蘭州: 蘭州大學, 2015.
[57] Singh P K, Negi A, Gupta P K,. Toxicophore exploration as a screening technology for drug design and discovery: Techniques, scope and limitations [J]., 2016, 90(8): 1785-1802.
[58] 何秋順. 基于結構特征的毒效因子發現 [D]. 廈門: 廈門大學, 2018.
[59] Wen B, Gorycki P. Bioactivation of herbal constituents: Mechanisms and toxicological relevance [J]., 2019, 51(4): 453-497.
[60] Kumar A, Zhang K Y J. Advances in the development of shape similarity methods and their application in drug discovery [J]., 2018, 6: 315.
[61] 唐玉煥, 林克江, 尤啟冬. 基于2D分子指紋的分子相似性方法在虛擬篩選中的應用 [J]. 中國藥科大學學報, 2009, 40(2): 178-184.
[62] 王山, 孫莉, 吳杰, 等. 一種基于計數型布隆過濾器的分子相似性算法研究 [J]. 計算機科學, 2017, 44(S2): 552-556.
[63] 吳萍, 孔德信. 分子相似性與MOLPRINT2D的本地化 [J]. 計算機與應用化學, 2008, 25(4): 505-508.
[64] Gong J Y, Cai C Q, Liu X F,. ChemMapper: A versatile web server for exploring pharmacology and chemical structure association based on molecular 3D similarity method [J]., 2013, 29(14): 1827-1829.
[65] He G Q, Song Y P, Wei W H,. eSHAFTS: Integrated and graphical drug design software based on 3D molecular similarity [J]., 2019, 40(6): 826-838.
[66] 溫聰聰. Me2Explorer: 一款應用于化學空間分析的集成軟件 [D]. 武漢: 華中農業大學, 2021.
[67] 段敬怡, 許妍妍, 王玉明, 等. 基于有害結局路徑的中藥毒性進程動態機制研究思路 [J]. 中國藥理學與毒理學雜志, 2020, 34(1): 58-63.
[68] Durai P, Ko Y J, Pan C H,. Evolutionary chemical binding similarity approach integrated with 3D-QSAR method for effective virtual screening [J]., 2020, 21(1): 1-18.
[69] Liu C X. Scientific understanding of toxicity and safety of Chinese medicines [J]., 2018, 10(2): 107.
Exploration and thinking of computer virtual screening technology in toxicity research of traditional Chinese medicine
QIAN Wen-xiu, YAN Xing-xu, ZHANG Wen-qing, JIA Guo-xiang, ZHAO Shan, SONG Li-li, LI Yu-bo
School of Traditional Chinese Medicine, Tianjin University of Traditional Chinese Medicine, Tianjin 301617, China
In recent years, people from all walks of life have paid more and more attention to the safety of traditional Chinese medicine (TCM), especially the toxicity research of TCM. Therefore, it is an urgent task to find an efficient technology to assist the toxicity research of TCM. The computer virtual screening technology has the advantages of high efficiency and convenience. After years of development, its technical theory has become mature and has been successfully applied in many fields. This article reviews the four main computer virtual screening technologies, such as molecular docking, machine learning, toxicophore and molecular similarity methods, and the application prospect of these technologies in the field of toxicity research of TCM are analyzed, aiming to provide reference for researchers engaged in toxicity research of TCM.
virtual screening; molecular docking; machine learning; toxicophore; molecular similarity; toxicity research of traditional Chinese medicine
R285
A
0253 - 2670(2023)12 - 4036 - 08
10.7501/j.issn.0253-2670.2023.12.029
2023-01-08
國家自然科學基金資助項目(81873194);國家中醫藥管理局青年岐黃學者支持項目
錢文秀,女,碩士研究生,研究方向為中藥學/毒性評價。E-mail: 13553195623@163.com
通信作者:宋麗麗,副教授,從事中藥分析與代謝組學研究。E-mail: sll0204@163.com
李遇伯,教授,博士生導師,從事中藥學/毒性評價研究。E-mail: yaowufenxi001@sina.com
[責任編輯 趙慧亮]
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
中老年保健(2021年4期)2021-12-01 11:19:40
中老年保健(2021年4期)2021-08-22 07:08:32
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
金橋(2020年7期)2020-08-13 03:07:00
數學物理學報(2020年2期)2020-06-02 11:29:24
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
