主動脈瓣膜間質細胞焦亡在鈣化性主動脈瓣疾病中的作用
2023-06-08 09:51:08宗麗娟杜占慧游福蓉王勤拯張成邢泉生
精準醫學雜志 2023年2期
關鍵詞:研究
宗麗娟 杜占慧 游福蓉 王勤拯 張成 邢泉生
(青島大學附屬婦女兒童醫院,山東 青島 266034 1 心臟中心;2 普外科)
隨著我國人民生活水平的提高以及人口老齡化程度的日益加劇,退行性鈣化性主動脈瓣疾病(calcific aortic valve disease,CAVD)在心臟瓣膜疾病中所占比例逐年上升,已成為臨床主動脈瓣置換的最主要病因[1-2]。CAVD以瓣膜發生纖維化和鈣化為特征,導致瓣葉增厚及啟閉功能受限,最終造成以心臟左室流出道梗阻為特征的心臟功能損害[3-4]。目前CAVD的治療方法包括藥物治療和經外科手術或經皮介入的主動脈瓣膜置換術[5]。其中藥物治療僅可緩解癥狀,無法控制疾病進展;而瓣膜置換術治療成本較高,術后人工瓣膜帶來的抗凝風險、瓣膜衰敗等問題仍困擾臨床[6]。因此研究CAVD的發生機制,確定導致CAVD進展的分子調控途徑,尋找潛在治療靶點,將為CAVD的治療提供新策略。
近年研究表明,CAVD是一種多細胞、多信號通路參與主動調控的疾病[7],其中作為主動脈瓣膜主體細胞的主動脈瓣膜間質細胞(AVICs),其炎性反應、成骨樣轉化、細胞死亡以及鈣鹽沉積等過程在CAVD的發生發展中發揮關鍵作用[8-12]。細胞焦亡是一種由焦亡蛋白Gasdermin家族介導的程序性細胞死亡方式,是機體的一種固有免疫反應,它既可以引起細胞的程序性死亡,同時又可導致周圍組織的炎癥反應。現已證實,細胞焦亡參與了多種疾病的發生發展[13-16],并與老年退行性疾病如老年性耳聾、阿爾茨海默病等發病密切相關[17],但與CAVD的發生是否相關至今尚未見報道。本研究通過體外培養AVICs并構建其鈣化模型,旨在探討細胞焦亡在CAVD中的作用。
1 材料與方法
1.1 主要試劑
杜爾貝克改良伊格爾(DMEM)培養基、胎牛血清購自美國Gibco公司,波形蛋白(Vimentin)、α-肌動蛋白(α-SMA)、Alexa Fluor 488標記羊抗兔IgG抗體購自英國Abcam公司,血小板-內皮細胞黏附分子(CD31)抗體購自美國ABclonal公司,Runt相關轉錄因子2(RunX2)、骨橋蛋白(OPN)、含Pyrin結構域NOD樣受體家族3(NLRP3)、焦亡素D蛋白(GSDMD)、半胱氨酸蛋白酶1(Caspase-1)、白細胞介素-1β(IL-1β)和IL-18購買于美國Proteintech公司,GSDMD-N和Cleaved-Caspase-1抗體購自美國CST公司,Ⅱ型膠原酶、β-磷酸甘油鈉、地塞米松、抗壞血酸及茜素紅染料購買于北京索萊寶有限公司,Caspase-1抑制劑VX-765購自美國MCE公司,細胞計數試劑盒(CCK-8)購自武漢三鷹生物技術有限公司。
1.2 AVICs的培養和鑒定
術中摘取主動脈瓣重度反流患者的主動脈瓣瓣葉后,立即放置于4 ℃的磷酸緩沖鹽溶液(PBS)中,轉運至實驗室。在無菌超凈臺中使用PBS清洗3次,無菌棉簽輕柔擦拭刮除瓣膜表面內皮細胞,使用眼科剪將瓣葉剪成約1 mm×1 mm大小的組織塊,置于PBS溶液配制的2 g/L的Ⅱ型膠原酶溶液中于37 ℃下消化6~7 h。將上述溶液1 000 r/min離心5 min,棄上清液,加入標準培養基[含體積分數0.12胎牛血清及100 kU/L青霉素/鏈霉素(1∶1)的DMEM培養基],在37 ℃、含體積分數0.05的CO2培養箱中培養至細胞融合度約達90%時進行傳代。用細胞免疫熒光法檢測細胞的間質細胞標志蛋白Vimentin、α-SMA及內皮細胞標志蛋白CD31以鑒定AVICs,使用熒光顯微鏡采集圖像。經過AVICs鑒定明確的第3~6代細胞可以用于后續的實驗。
1.3 VX-765溶液有效濃度的篩選
為篩選VX-765有效干預濃度,利用CCK-8實驗檢測VX-765對AVICs的細胞毒性。將處于對數生長期的AVICs接種于96孔板中,待細胞生長24 h且完全貼壁后,分別加入含有不同濃度(0、1、2.5、5、10、20、40、60、80 μmol/L)的VX-765標準培養基,每種濃度設置6個復孔。將孔板置于37 ℃、含體積分數0.05的CO2培養箱中培養24 h以后每孔加入CCK-8溶液10 μL,將孔板在培養箱中孵育2 h以后,使用酶標儀測量各濃度組細胞在450 nm波長處的吸光度(A)值,計算細胞存活率。細胞存活率=[(A實驗孔-A空白孔)/(A對照孔-A空白孔)]×100%,選擇干預藥物的最佳濃度用于后續實驗。
1.4 分組及處理
將傳代細胞按照每孔約1.2×106個接種于6孔板中,培養24 h后。依據處理方式不同將細胞分為3組,對照組以標準DMEM 培養基培養14 d,鈣化組以鈣化培養基(含10 mmol/L β-磷酸甘油鈉+100 nmol/L地塞米松+50 mg/L抗壞血酸的標準DMEM培養基)培養14 d,而抑制劑組則使用含10 μmol/L VX-765的DMEM培養基預處理24 h后轉入鈣化培養基繼續培養14 d。
1.5 AVICs中鈣結節表達水平檢測
將各組細胞棄培養基,用40 g/L多聚甲醛溶液固定,經去離子水清洗以后加入茜素紅染液,染色5 min后棄去染液,去離子水洗滌終止反應,普通光學顯微鏡下觀察鈣鹽沉積情況。
1.6 AVICs中鈣化、焦亡相關蛋白及炎癥因子表達水平檢測
將培養好的細胞接種于6孔板,待AVICs細胞融合度約達50%時,按1.4步驟處理細胞14 d,收集AVICs細胞并提取細胞中總蛋白,加入5×loading buffer,并置于水中煮沸15 min進行變性,保存于-80 ℃環境下。按照SDS-PAGE凝膠配置說明書配制分離膠和濃縮膠,加15 μg蛋白樣品至凝膠孔中進行SDS-PAGE蛋白電泳,PVDF轉膜之后以50 g/L脫脂奶粉室溫封閉2 h。根據目的蛋白分子量裁剪PVDF膜條帶,并分別加入鈣化相關蛋白OPN、RunX2,細胞焦亡相關蛋白NLRP3、GSDMD、Caspase-1、GSDMD-N、Cleaved-Caspase-1,炎癥因子IL-1β、IL-18以及內參β-actin單克隆抗體,4 ℃孵育過夜。以β-actin作為內參照,TBST洗3次,每次10 min,加入二抗并室溫孵育1 h,TBST洗膜3次,每次10 min,最后加入ECL化學發光液顯影并拍照。使用Image J 7.0軟件分析上述蛋白條帶灰度值,目標蛋白相對表達量以目標蛋白灰度值/β-actin蛋白灰度值計算。實驗設置6個復孔,重復測量3次,結果取均值。
2 結 果
2.1 AVICs細胞表型鑒定
免疫熒光染色鑒定AVICs細胞表型,結果顯示近乎所有細胞均可見間質細胞標志蛋白Vimentin及α-SMA表達,未見內皮細胞標志蛋白CD31表達(圖1),說明分離培養的AVICs純度理想,無明顯內皮細胞污染,可以用于后續實驗。
2.2 AVICs體外鈣化模型的建立
茜素紅染色結果顯示,鈣化組細胞呈現橙色著色,對照組細胞無明顯陽性著色(圖2)。蛋白免疫印跡檢測結果顯示,鈣化組與對照組當中鈣化相關蛋白OPN的相對表達量分別為1.28±0.16、0.32±0.03,RunX2的相對表達量則分別為2.27±0.15、0.39±0.02,鈣化組中鈣化相關蛋白OPN及RunX2的相對表達量均顯著高于對照組(t=6.568、12.204,P<0.01)。見圖3。
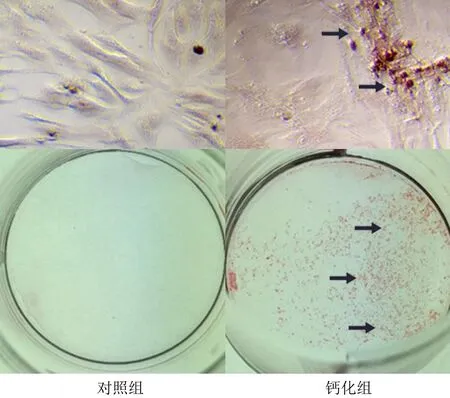
箭頭所指區域為鈣化結節
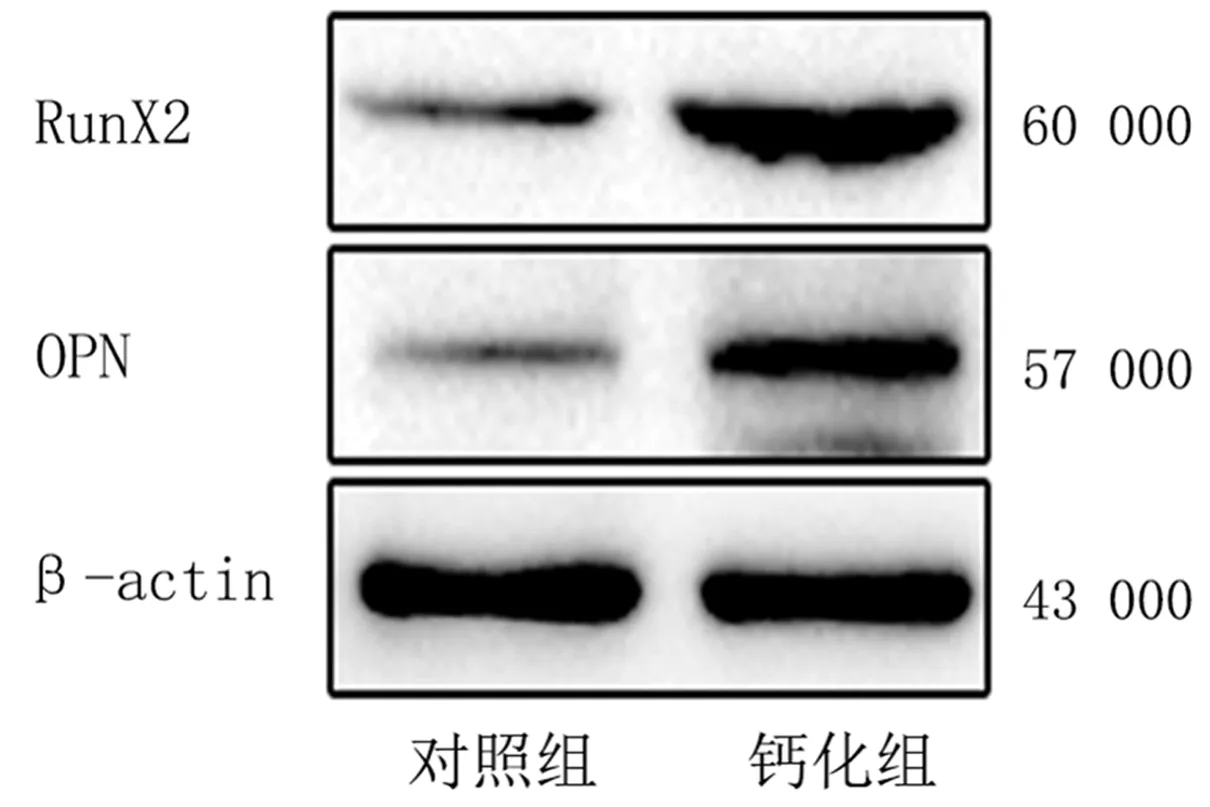
圖3 對照組和鈣化組AVICs中鈣化相關蛋白表達水平
2.3 對照組和鈣化組中AVICs焦亡相關蛋白表達水平比較
免疫印跡法檢測結果顯示,鈣化組焦亡相關蛋白NLRP3、GSDMD、Caspase-1、GSDMD-N以及Cleaved-Caspase-1相對表達量顯著高于對照組(t=4.586~15.842,P<0.01)。見圖4、表1。
表1 對照組和鈣化組AVICs焦亡相關蛋白的表達水平比較
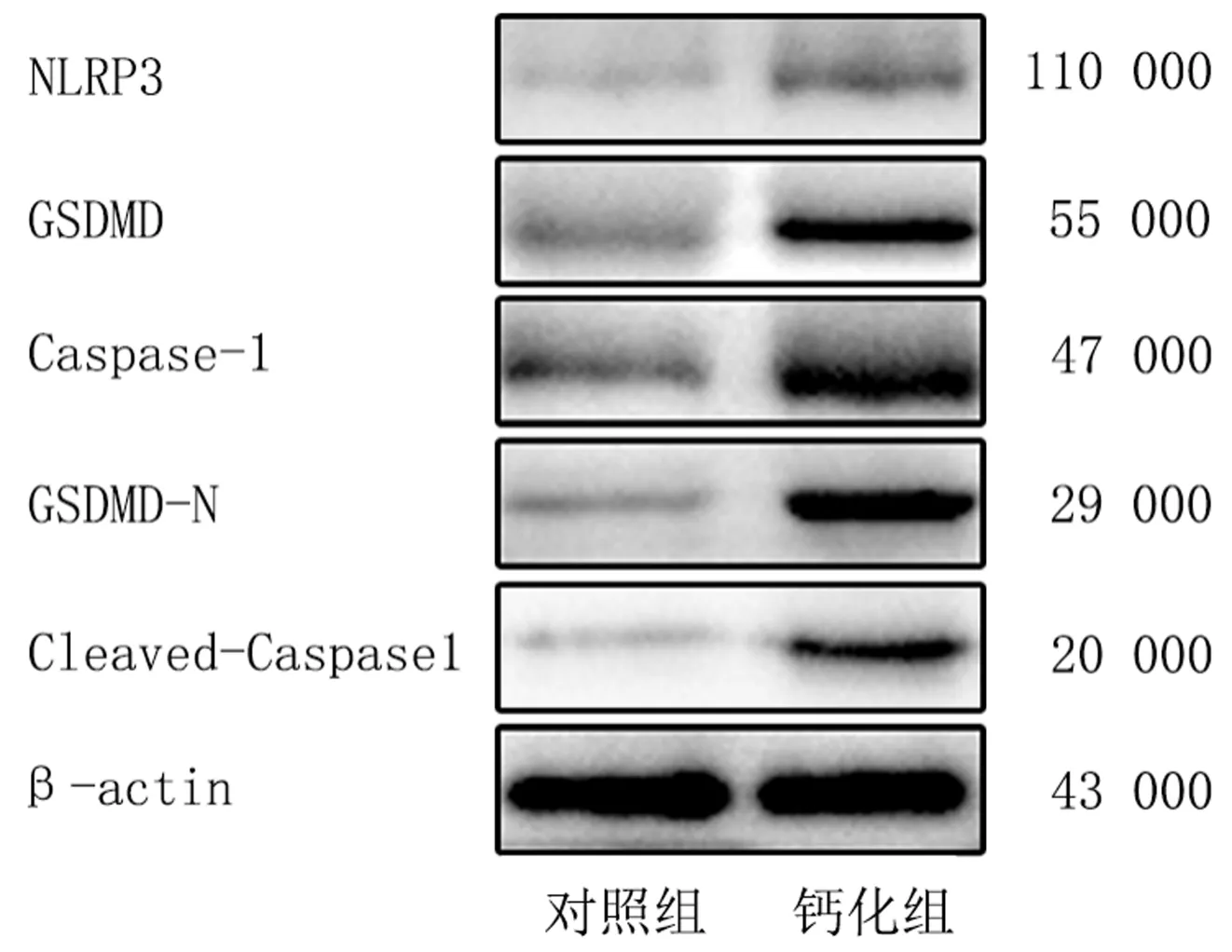
圖4 對照組和鈣化組AVICs焦亡相關蛋白表達水平
2.4 VX-765干預AVICs最佳濃度確定
CCK-8法檢測結果顯示,在VX-765濃度為0、1、2.5、5、10、20、40、60、80 μmol/L的標準培養基中,AVICs的存活率分別為(92.407±3.431)%、(95.725±1.203)%、(99.642±1.877)%、(100.750±6.751)%、(101.975±7.023)%、(87.975±1.141)%、(88.742±1.553)%、(83.650±1.648)%、(72.817±5.514)%,與濃度為10 μmol/L相比,當VX-765濃度為20、40、60、80 μmol/L時AVICs的存活率均顯著降低(t=2.200~5.631,P<0.05),因此后續實驗中抑制劑組均采用含10 μmol/L VX-765的標準培養基預處理AVICs 24 h后,再進行后續鈣化培養基培養。
2.5 鈣化組與抑制劑組中AVICs鈣化、焦亡相關蛋白及炎性因子表達水平比較
茜素紅染色結果顯示,與鈣化組相比,抑制劑組橙紅色顆粒減少,鈣化結節消退(圖5)。免疫印跡法結果顯示,抑制劑組OPN、RunX2、NLRP3、GSDMD、Caspase-1、GSDMD-N、Cleaved-Caspase-1以及IL-1β、IL-18的相對表達量均顯著性低于鈣化組(t=5.799~19.236,P<0.01)。見圖6、表2。
表2 鈣化組與抑制劑組AVICs鈣化、焦亡相關蛋白及炎性因子表達水平比較
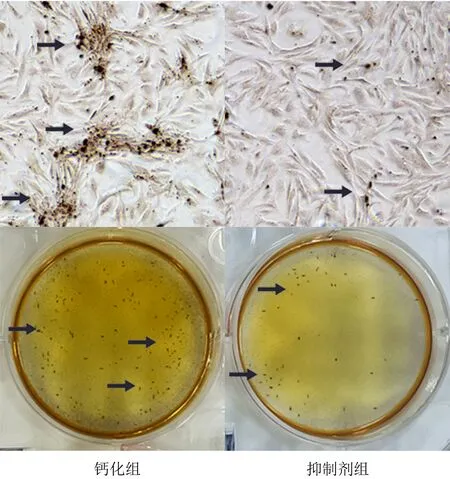
箭頭所指區域為鈣化結節
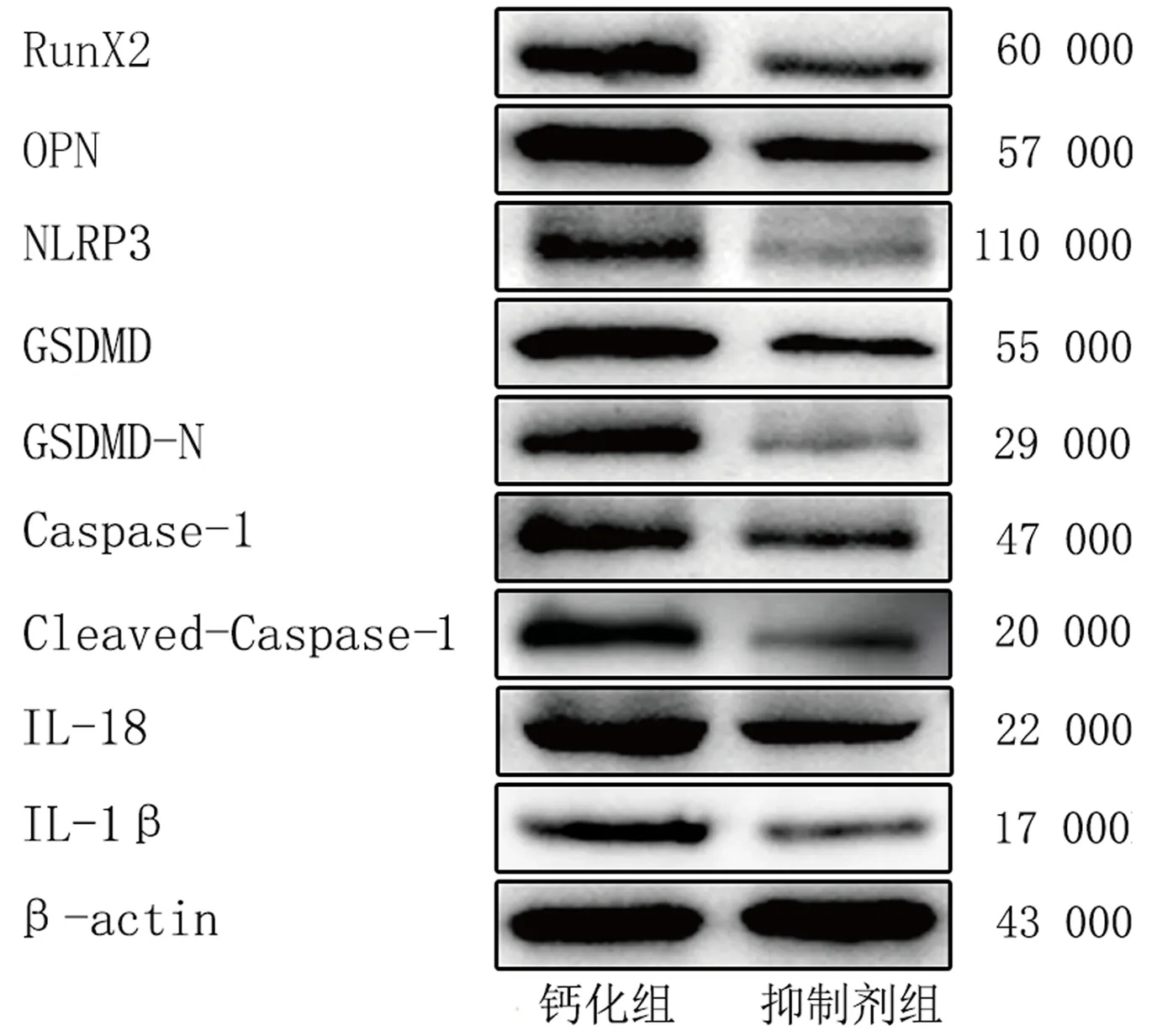
圖6 鈣化組與抑制劑組AVICs鈣化、焦亡相關蛋白及炎癥因子表達水平
3 討 論
近年來,CAVD的發病率在老年人群中呈增長趨勢,隨著瓣膜病變程度加重,該病逐漸破壞老年患者血流動力學穩定性,發展至后期極易危及患者生命。雖然已經有大量關于CAVD的病因學及病理生理學方面的研究,但是CAVD的主要發病機制依然不明確。CAVD涉及到主動脈瓣瓣葉進行性增厚、基質重塑以及鈣鹽沉積等過程,上述過程導致瓣葉嚴重鈣化、結構和功能減退,最終發生主動脈瓣嚴重狹窄和(或)關閉不全[18]。AVICs是主動脈瓣的主要組成成分之一,且AVICs向成骨樣細胞分化的過程在CAVD疾病發展中起主要作用,這也是目前探討CAVD發生機制的主要研究方向。多項研究表明,CAVD中主動脈瓣鈣化早期病變與血管鈣化相似,是一個異位鈣化的過程,其中成骨細胞激活與間質細胞死亡同時存在。細胞焦亡屬于細胞程序性壞死的一種,在動脈粥樣硬化、血管鈣化等多種心血管疾病進程中發揮重要作用[19],但CAVD中是否存在細胞焦亡及其相關機制目前尚未有研究報道。
本研究使用鈣化培養基,建立AVICs體外鈣化模型并進行驗證。結果顯示經過鈣化培養基誘導的AVICs呈聚集樣生長,茜素紅染色呈現橙色著色,證明AVICs中存在鈣鹽沉積。免疫印跡法檢測發現,鈣化相關蛋白OPN和RunX2表達增多,提示AVICs向成骨樣細胞分化。在此模型上,本研究進行了細胞焦亡的相關檢測。細胞焦亡是由GSDMD介導的一種新型程序性炎癥性細胞死亡模式,其特征是細胞腫脹變大,膜上孔洞形成及炎癥因子等細胞內容物的釋放。根據活化蛋白不同,細胞焦亡分為依賴Caspase-1的經典途徑和依賴Caspase-4/5/11的非經典途徑,目前經典激活途徑研究較多[20]。在經典途徑細胞焦亡中,Caspase-1作為焦亡的效應分子,通常以pro-Caspase-1的形式存在于細胞中,在各種損傷因素的刺激下,NLRP3等炎癥小體被激活,并將pro-Caspase-1裂解成Caspase-1,活化的Caspase-1隨后將GSDMD裂解為GSDMD-N,釋放出的GSDMD-N在細胞膜上形成孔洞,進而促進炎性因子的釋放和細胞腫脹,激活細胞焦亡[21]。細胞焦亡在機體內是一個動態過程,往往難以觀測,目前活化的Caspase-1和GSDMD-N已被公認為細胞焦亡發生的標志物,通過檢測這些指標的水平可以確定細胞焦亡的發生情況。本研究中AVICs經鈣化培養基誘導以后,焦亡相關蛋白NLPR3、GSDMD、Caspase-1、GSDMD-N、Cleaved-Caspase-1等表達升高,表明AVICs向成骨樣細胞改變的過程中存在細胞焦亡發生。
為進一步明確細胞焦亡在AVICs成骨樣改變中的作用,本研究使用VX-765抑制焦亡效應分子Caspase-1的活性。VX-765是一種具有口服生物活性的Caspase-1抑制劑,現已被應用于銀屑病和癲癇患者的Ⅱ期臨床試驗。本研究使用VX-765干預鈣化培養基誘導的AVICs,通過CCK-8法確定VX-765的干預時間及濃度。實驗結果顯示,VX-765干預減少了細胞內鈣結節的形成,且VX-765抑制劑組中的鈣化相關蛋白OPN、RunX2,焦亡相關蛋白NLPR3、GSDMD、Caspase-1、GSDMD-N、Cleaved-Caspase-1及炎癥因子IL-18、IL-1β的相對表達量均顯著降低,上述結果提示VX-765能夠減少細胞焦亡的發生,抑制AVICs向成骨樣細胞分化。
細胞焦亡在本質上是一種炎癥過程,而炎癥在CAVD進展過程中發揮重要作用[22]。多項研究表明,慢性炎癥反應貫穿瓣膜鈣化的全過程,炎癥信號通路的激活、炎性因子的表達和釋放在AVICs向成骨樣細胞分化中扮演重要角色[18,23]。在細胞焦亡中,NLPR3/Caspase-1通路的激活是焦亡的關鍵過程。已有研究表明,炎小體在多種衰老相關的疾病中發揮關鍵作用,可以作為心血管疾病治療的重要靶點[24]。有研究發現,咖啡酸苯乙酯可以通過抑制Akt/NF-κB通路和NLPR3炎癥小體的激活來減少條件培養基誘導下AVICs鈣化結節的形成[25]。根據本研究結果,我們推測VX-765可能通過靶向抑制Caspase-1來減少細胞焦亡的發生,從而減輕炎癥反應,延緩AVICs的成骨樣分化。
綜上所述,本研究結果表明在AVICs鈣化及成骨樣分化過程中存在細胞焦亡,應用VX-765干預后可有效緩解鈣化培養基誘導下AVICs鈣化結節的形成,其機制可能為VX-765干擾細胞焦亡中炎癥小體形成,減少AVICs成骨樣分化。因此,進一步研究細胞焦亡在CAVD中的作用機制將為臨床防治CAVD提供新的作用靶點和治療策略。
倫理批準和知情同意:本研究涉及的所有試驗均已通過青島大學附屬婦女兒童醫院醫學倫理委員會的審核批準(文件號QFELL-YJ-2020-80)。所有試驗過程均遵照《赫爾辛基人體研究倫理準則宣言》的條例進行。受試對象或其親屬已經簽署知情同意書。
作者聲明:宗麗娟、邢泉生、杜占慧、游福蓉、王勤拯參與了研究設計;宗麗娟、杜占慧、邢泉生、張成參與了論文的寫作和修改。所有作者均閱讀并同意發表該論文。所有作者均聲明不存在利益沖突。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車工程學報(2017年2期)2017-07-05 08:13:02
國際商務財會(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
