鈉-牛磺膽酸共轉運多肽缺陷患兒的肝功能及基因特點
2022-12-23 07:33:04孫雁楠朱莉
貴州醫科大學學報 2022年12期
孫雁楠, 朱莉
(1.貴州醫科大學 臨床醫學院, 貴州 貴陽 550000; 2.貴陽市婦幼保健院 兒童消化科, 貴州 貴陽 550000)
鈉-牛磺膽酸共轉運多肽(sodium taurocholate cotransporting polypeptide,NTCP)缺陷病是近幾年新發現的一種遺傳性膽汁酸代謝疾病,是溶質轉運蛋白家族10成員(solute carrier family 10 member 1,SLC10A1)雙等位基因突變導致NTCP功能障礙[1]。研究顯示NTCP缺陷與新生兒高膽紅素血癥及嬰兒早期膽汁淤積癥有關[2-4]。NTCP位于肝細胞基底外側膜上,可將血漿結合型膽汁酸鹽轉運至肝臟,當NTCP功能異常時肝臟攝取膽汁酸障礙,血液中膽汁酸堆積,引起高膽汁酸血癥為主要表現的血清學指標異常,已發現多個突變位點可促成該病[5-6]。NTCP缺陷病在兒童和成人中均可發病,共同特征均為血漿中持續升高的膽汁酸[2-3,7],該病于2015年首次報道[7],國內外報道案例散發[1-4,6-13],對本病的遺傳學特征、臨床表現、治療及遠期預后尚無統一定論,需要更多研究和報告進一步充實,因此本研究對經基因檢測確診的11例NTCP缺陷患兒肝功能及基因檢測結果進行分析,報告如下。
1 對象與方法
1.1 研究對象
選取2019—2021年經高通量基因測序確診的NTCP缺陷病患兒作為研究對象,要求年齡≤14歲,膽汁酸升高為主,伴有或不伴有其他肝功能指標異常,遺傳代謝性基因檢測完善及院外未進行治療。排除標準:(1)病例資料不完善;(2)解剖結果為異常、感染、藥物、靜脈營養等所致肝功能異常。共納入NTCP缺陷病患兒11例,男性6例(54.5%)、女性5例(45.5%),同時選取患兒父親11人、母親11人作為參照,行高通量基因測序。本研究獲得醫院倫理委員會批準(2022-39)及患兒監護人簽署知情同意書。
1.2 研究方法
1.2.1一般臨床資料 收集NTCP缺陷病患兒的年齡、性別、主訴、出生史、胎產次、喂養史、家族史及新生兒期黃疸情況等一般臨床資料。
1.2.2肝功能 抽取患兒入院治療前清晨空腹靜脈血5 mL,采用西門子全自動化分析儀[西門子醫學診斷產品(上海)有限公司]及相應試劑盒檢測血清谷丙轉氨酶(alanine aminotransferase,ALT;參考值0~40 U/L)、谷草轉氨酶(aspartate aminotransferase,AST;參考值0~40 U/L)、總膽紅素(total bile acid,TBIL;參考值0~17 μmol/L)、直接膽紅素(direct bilirubin,DBIL;參考值0~5.1 μmol/L)、間接膽紅素(indirect bilirubin,IBIL;參考值0~13.7 μmol/L)、膽汁酸(total bile acid,TBA;參考值0~10 μmol/L)。出院后定期于門診隨診復查上述指標。
1.2.3高通量測序 取患兒及父母外周血3 mL,置于乙二胺四乙酸(ethylenediaminetetraacetic acid,EDTA)抗凝管中送至北京麥基諾基醫學檢驗公司進行遺傳代謝性肝病573個基因panelV2檢測,目標基因片段通過第2代高通量測序儀進行測序;突變基因依據美國醫學會遺傳學與基因組學會(american college of medical genetics and genomics,ACMG)發布的變異解讀指南進行致病性分析。
1.2.4危害性預測 發現明確或可疑與臨床表型相關的基因突變時,對患兒突變基因與其父母基因進行Sanger驗證,并運用Rare Exome Variant Ensemble Learner(REVEL)、Sortig Intolerant from Tolerant(SIFT)、PolyPhen-2、Mutationtation Taster、Genomic Evolutionary Rate Profiling(GERP+)5種生物信息學蛋白功能綜合性預測軟件對突變位點進行危害預測。
1.2.5隨訪 出院后于消化科門診隨訪1年,每次復診當日空腹抽取靜脈血5 mL進行ALT、AST、TBA、TBIL、DBTL及IBIL等肝功能指標檢測。
2 結果
2.1 一般情況
11例基因檢測確診的NTCP缺陷病患兒男性6例(54.5%)、女性5例(45.4%),男女比例1.2 ∶1,就診年齡3~219 d、平均(60.37±71.20)d,確診年齡40~221 d、平均(99.82±69.63)d,混合喂養5例、人工喂養1例及母乳喂養5例,因黃疸就診3例、腹瀉就診1例、體檢發現肝功能異常就診2例、當地新生兒科住院發現TBA升高轉診本院5例,患兒直系親屬中有肝衰竭病史1例、母親孕期合并高TBA血癥1例、余9例家族史無特殊,新生期出現黃疸4例(其中有1例診斷新生兒ABO溶血)、7例無黃疸。
2.2 肝功能情況
11例NTCP缺陷病患兒中ALT升高占比72.7%(8/11),AST升高占比72.7%(8/11),TBA升高占100%(11/11),TBIL升高占比54.5%(6/11),DBIL升高占比63.6%(7/11),IBIL升高占比36.4%(4/11);NTCP缺陷病患兒都存在單項或多項肝功能指標異常,但都存在TBA升高;肝功能指標除TBA外,余各項指標在隨訪過程中呈下降趨勢(病例1、5患兒失訪),隨訪4~8周后,ALT、AST、TBIL、DBIL及IBIL恢復正常有5、4、2、1及2例,TBA在治療后一段時間內仍呈高水平,但隨著隨訪時間延長TBA總體呈下降趨勢。見表1和圖1。
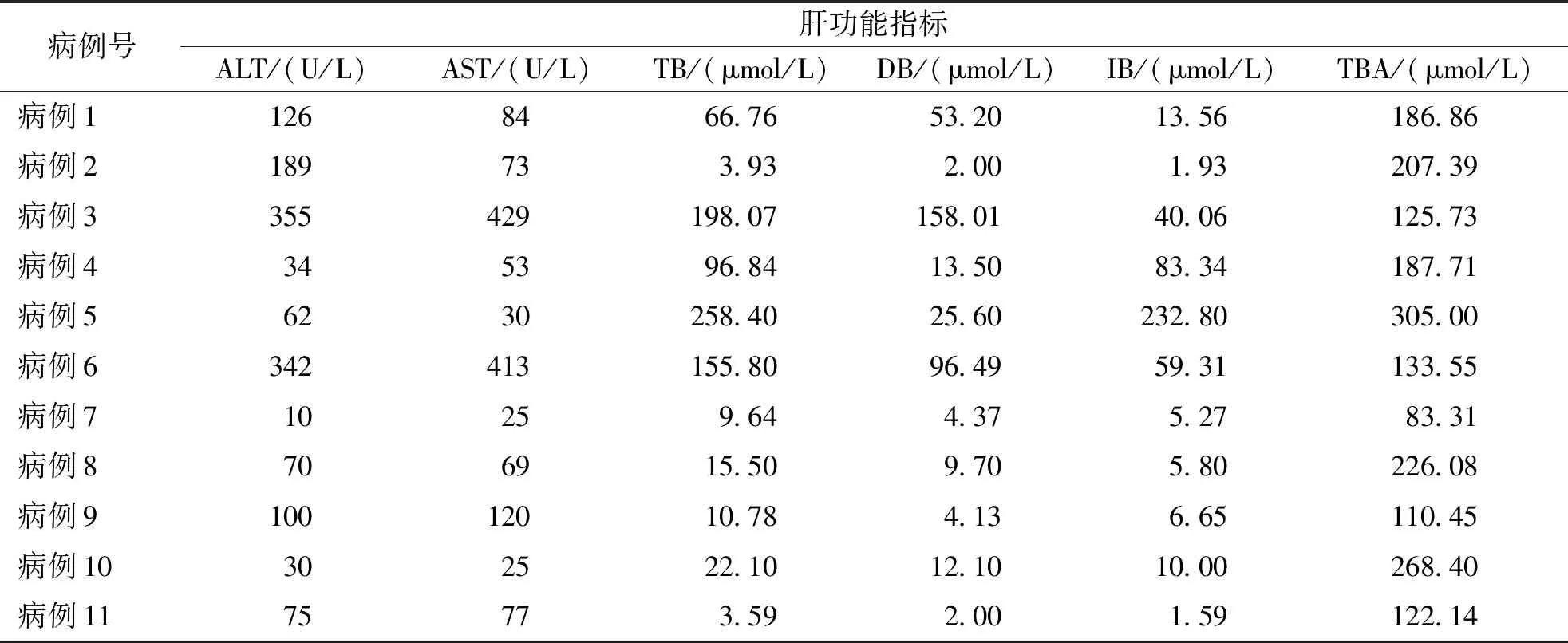
表1 NTCP缺陷病患兒治療前的肝功能指標Tab.1 Pre-treatment liver function indicators in children with NTCP deficiency

注:A~F分別為ALT、AST、TBA、TBIL、DBIL及IBIL隨訪情況;P表示病例,數字為病例順序號,其中P1、P5表示失訪。圖1 NTCP缺陷病患兒隨訪期間的肝功能變化Fig.1 Changes in liver function during follow-up of children with NTCP deficiency disease
2.3 基因分析
11例NTCP缺陷病患兒基因測序結果顯示,染色體chr14:70245193上SLC10A1基因均存在相同突變位點,系第800號核苷酸由胞嘧啶C變為胸腺嘧啶T(c.800C>T)的純合突變,上述突變導致第267號氨基酸由絲氨酸變為苯丙氨酸(p.S267F),屬于錯義突變;與父母基因比對,結果顯示11例NTCP缺陷病患兒基因突變均來源于父母雙方,父母均為SLC10A1基因(c.800C>T)突變攜帶者。見圖2。
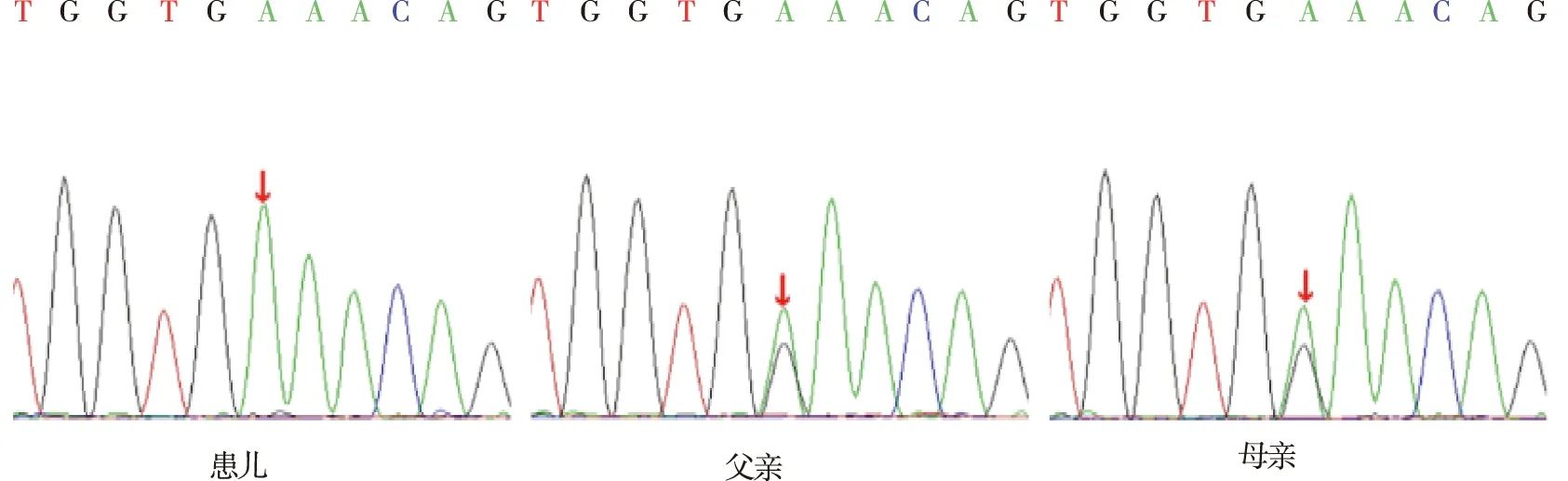
注:紅色曲線表示胸腺嘧啶T,黑色曲線表示鳥嘌呤G,綠色曲線表示腺嘌呤A,藍色曲線表示胞嘧啶C;紅色箭頭表示變異位點定位。圖2 病例1號與父母SLC10A1基因的Sanger驗證Fig.2 Sanger validation of SLC10A1 gene between case 1 and parents
2.4 突變位點危害性的生物信息學預測
所有NTCP缺陷病患兒SLC10A1基因(c.800C>T)經REVEL軟件預測結果為良性,經SIFT、P0lyPhen-2、Mutationtation Taster、GERP+預測結果均為有害。
2.5 其他變異信息
11例NTCP缺陷病患兒除檢測到與疾病表型高度相關的SLC10A1基因(c.800C>T)突變外,還檢測到與臨床相關性不明確位點134個,這些基因與肝豆狀核變性、高密度脂蛋白缺乏病、Gilbert綜合征等多種疾病有關,其中UGT1A1基因(c.211G>A,p.G71R)在11名患兒中出現頻率最高(4/11),該基因突變表型與Gilbert綜合征、Crigler-Najjar綜合征、家族性暫時性新生兒高膽紅素血癥有關,且以黃疸為主要表現、IBIL升高為主、通常不伴TBA的升高,但本研究中攜有UGT1A1基因(c.211G>A)突變的病例中,2例有黃疸表現,但血生化以DBIL升高為主,且TBA明顯升高,故與患兒的臨床表型不相符。見表2。
表2 NTCP缺陷病患兒與臨床相關性不明確的突變位點結果Tab.2 Results of mutation loci with unclear clinical relevance in children with NTCP defect
注:—表示未檢測出。
3 討論
NTCP是肝細胞發揮Na+依賴吸收功能的主要轉運蛋白,轉運結合與非結合性TBA,在TBA的轉運過程中發揮關鍵性作用,對前者更具有親和力[14]。研究顯示,NTCP還可作為乙肝及丁肝病毒進入肝細胞的受體,人類SLC10A1基因突變使NTCPTBA轉運功能障礙,血中TBA升高導致NTCP缺陷病的發生[15-16];另一方面SLC10A1基因突變可消除甲型肝炎病毒(hepatitis A virus,HAV)或丁型肝炎病毒(hepatitis D virus,HDV)感染肝細胞的能力[17]。本研究11例確診NTCP缺陷病患兒,確診年齡40~221 d,男孩多于女孩。關于NTCP缺陷病的臨床表現報道存在差異,Vaz等[7]研究顯示NTCP缺陷病例表現為肌張力減低;Liu等[18]研究則提示為25(0H)D缺乏;孫文君等[8]研究顯示2例患兒中1例黃疸,血清25(OH)D缺乏,1例表現為生長發育落后。本研究因黃疸首次就診者3例,肝功能指標以TBA、ALT、AST、DBIL及TBIL升高為主,臨床表現、實驗室檢查與肝細胞性黃疸患者相似,入院初診為嬰兒膽汁淤積性肝病;另有部分患兒因其他系統疾病或體檢發現TBA升高,無瘙癢等高TBA中毒表現,臨床上易漏診。研究顯示NTCP缺陷與新生兒期高膽紅素血癥和部分嬰兒早期的膽汁淤積有關[2-4]。發生機制與 NTCP缺陷病在肝細胞基底側膜上的另一種轉運蛋白有機陰離子轉運多肽1B1(organic anion transporting polypeptide 1B1,OATP1B1)/有機陰離子轉運多肽1B3(organic anion transporting polypeptide 1B3,OATP1B3)的代償作用有關,OATP1B1/OATP1B3蛋白特異性底物范圍廣泛,能將血漿中的非結合TBA、膽紅素、內源性激素、外源性毒性物質攝入肝臟[19],緩解NTCP缺陷導致的高TBA環境,但是NTCP缺陷病患兒明顯升高的血漿TBA又可競爭性抑制OATP1B1/OATP1B3攝取膽紅素的功能,導致膽紅素繼發性升高[9-10]。
目前NTCP缺陷病經報道的SLC10A1基因突變位點有c.(800.C>T)、c. 755G >A (p.Arg252His)、c.615-618del(p.Ser206Profs*12)、c.263T>C(p.lle88Thr)及c.595A>C(p.Ser199Arg),近期國內還報道了2種新的致病突變c.374dupG和c.682_683delCT,目前SLC10A1突變基因譜在不斷更新[1,4,7,11]。自2015年國外報道首例NTCP缺陷病兒童病例以來,截至目前,我國共報道了80余例NTCP缺陷病兒童[1,5,9,12-13,18,20],約94.5%的患兒SLC10A1上存在c.800C>T純合突變[21]。本研究11例NTCP缺陷患兒均為SLC10A1 c.800C>T(p.Ser267Phe)純合突變,其父母均為c.800C>T(p.Ser267Phe)的突變基因攜帶者,表明c.800C>T(p.Ser267Phe)是SLC10A1高頻突變位點。目前報道的病例大部分為純合突變,少部分為雜合突變,Dong等[20]研究中13例NTCP缺陷兒童中c.800C>T(p.Ser267Phe)純合突變12例,c.812A>G(p.Asn271Ser)復合雜合突變僅1例。
NTCP缺陷病目前尚缺乏確切的治療手段,臨床以對癥治療為主[3]。本研究結果顯示,NTCP缺陷病患兒肝功能指標呈單項或多項指標異常,TBA升高為共同特征。根據患兒具體情況,個體化的選擇治療方案:當嚴重肝酶升高時,使用保肝藥物復方甘草酸苷、還原型谷胱甘肽、葡醛內酯等聯合熊/鵝去氧膽酸、消膽胺等利膽藥物治療;當膽汁淤積時使用保肝、利膽藥物,予高三酰甘油配方奶粉喂養,口服益生菌、補充脂溶性維生素并聯合使用利膽藥物治療。此外,本研究NTCP缺陷病患兒門診隨訪結果提示:肝酶、膽紅素等指標在短期內降至正常,TBA波動幅度較大,但最終呈下降趨勢,總體治療效果良好。有研究指出OATP1B1/OATP1B3蛋白表達量可隨年齡增長而增加,推測其對NTCP缺陷的代償作用隨年齡增長而增強[22]。同時,生后1歲時分子翻譯后的糖基化過程對于NTCP在肝細胞膜基底側的正確定位至關重要,推測NTCP缺陷病患兒TBA可能不會無限升高,可能隨著年齡增長逐漸改善[23]。
關于NTCP的表達與調控,法尼醇受體及其他核受體使肝TBA轉運蛋白發生適應性轉錄調節,下調NTCP、減少TBA攝取,并誘導替代性基底外側流出蛋白有機溶質轉運體α/β(organic solute transporter α/β,OSTα/β)、多重耐藥相關蛋白3(multidrug resistant associated 3,MRP3)等促進TBA外排,緩解高濃度TBA對肝細胞的損害[24-25]。此外,國外研究表明,牛磺石膽酸(taurolithocholic acid,TLC)可持久抑制NTCP蛋白的表達,而環磷酸腺苷(cyclic adenosine monophosphate,cAMP)能通過促使容納NTCP蛋白的細胞囊泡進入肝細胞膜迅速上調NTCP蛋白的表達[26]。
NTCP缺陷病共同特征為頑固性高TBA血癥,由于部分兒童病例表現為膽汁淤積使黃疸成為相對常見的NTCP缺陷病首診原因,而成人病例中大都無癥狀,少部分育齡期女性表現為妊娠期肝內膽汁淤積癥,導致剖宮產率的增加[27]。因缺乏陽性癥狀導致本病識別困難,可能是目前NTCP缺陷病報道較少的原因之一。
目前隨著對NTCP分子機制深入的研究,不斷有新的變異位點被發現,豐富了SLC10A1基因變異譜,引起人們對本病的更多的重視;目前尚未出現有關本病導致肝硬化、肝衰竭等終末期肝病的報道,結合其代償機制OATPlBI/OATPlB3表達量呈年齡依賴性增加的特點,關于NTCP缺陷病是否需要積極治療還存在疑問,對其遠期預后的判斷可能需要一個較長的觀察隨訪過程。
猜你喜歡
肝博士(2024年1期)2024-03-12 08:38:08
肝博士(2022年3期)2022-06-30 02:48:58
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
汽車工程學報(2017年2期)2017-07-05 08:13:02
中國衛生標準管理(2015年1期)2016-01-14 03:41:20
中國藥業(2014年12期)2014-06-06 02:17:26
