MOF-74負載磷鎢酸催化劑的制備及其催化環己烯氧化制己二酸性能
2022-12-15 11:38:30史慶偉王璐璐王吉林
石油煉制與化工 2022年12期
史慶偉,王璐璐,王吉林
(遼寧石油化工大學石油化工學院,遼寧,撫順 113001)
己二酸是一種重要的有機化工原料,在尼龍、醫藥、農藥、染料、食品添加劑等領域有重要應用前景。目前工業上生產己二酸的方法主要是環己醇/酮硝酸氧化法和環己烯水合硝酸氧化法[1]。但是,這兩種合成方法具有過程復雜、設備腐蝕嚴重,以及反應過程釋放N2O氣體,破壞大氣層,導致環境污染等問題。因此,人們急需要尋找一種環境友好的反應路徑來代替上述方法。綜合文獻分析發現,開發新型的、可直接氧化環己烯制己二酸的催化劑是一種比較有潛力的解決辦法。在已經報道的催化劑中,磷鎢酸(H3PW12O40,簡稱PTA)有很高的環己烯氧化制己二酸的催化活性,但是PTA催化劑存在穩定性差、反應結束后催化劑回收困難等缺點[2],限制了其進一步推廣使用。雖然通過將PTA負載在分子篩[3]、二氧化硅[4]、金屬化合物[5]等載體上的手段可以緩解上述問題,但常常會衍生出PTA單純依賴物理吸附、難以穩定在載體結構中、容易從載體流失、催化劑結塊團聚、催化活性不高等諸多新問題。
近年來,金屬有機骨架材料(MOFs)作為一類具有網絡拓撲結構的新型多孔材料在催化劑領域發揮了重要作用,其特殊的孔道結構限制和穩定了活性組分,可以進一步促進產物的分離和催化劑的回收。因此,將活性組分負載在MOFs的孔道中是一種有效的、可以改善傳統負載型催化劑存在問題的途徑。在眾多MOFs材料中,M-MOF-74(M為Mg,Zn,Cu,Mn等二價過渡金屬)具有孔徑大、比表面積大、結構穩定且具有不飽和金屬活性位點等優點而受到廣泛關注[6-7]。
本研究以Cu2+為無機活性金屬中心、2,5-二羥基對苯二甲酸為有機配體、PTA為活性組分,采用熱溶劑法一步原位制備適合環己烯氧化制己二酸的催化劑,以期通過將PTA負載于M-MOF-74的骨架中,有效解決單純PTA催化劑易流失、易腐蝕設備和傳統負載型催化劑PTA易結塊團聚,以及催化活性不高等問題。
1 實 驗
1.1 主要試劑
三水合硝酸銅,2,5-二羥基對苯二甲酸,PTA,N,N-二甲基甲酰胺(DMF),無水甲醇,環己烯,雙氧水(H2O2質量分數為30%),均為分析純,國藥集團化學試劑有限公司提供。
1.2 x%PTA@Cu-MOF-74催化劑制備
稱取三水合硝酸銅0.731 g(3.028 mmol)、2,5-二羥基對苯二甲酸0.299 g(1.514 mmol)置于100 mL燒杯中,加入DMF 54 mL 和無水甲醇6 mL,同時加入一定質量的PTA,超聲波振蕩攪拌,直至溶液混合均勻。然后轉移至100 mL內襯聚四氟乙烯的反應釜中,置于烘箱120 ℃晶化24 h。反應結束后,冷卻至室溫,過濾,將上層清液倒出,洗滌下層固體。具體洗滌過程為:先用DMF洗滌,充分攪拌除去未反應的配體等雜質,再用無水甲醇洗滌置換出孔道內的DMF,過濾,重復上述步驟至少3次。最后置于烘箱中,在100 ℃下真空干燥12 h,制得PTA負載量(w)為x%的PTA/Cu-MOF-74催化劑,記作x%PTA@Cu-MOF-74(x=0,10,20,30,40),裝袋備用。其中,PTA負載量(w)為PTA占催化劑的質量分數。
1.3 環己烯催化氧化反應
在裝有冷凝管和溫度計的三口燒瓶中進行環己烯催化氧化反應。先將一定量的催化劑和過氧化氫于三口燒瓶中均勻混合,然后向上述溶液中緩慢滴加環己烯,升溫到預定溫度,反應特定時間后,停止加熱,自然冷卻,過濾分離催化劑,將濾液置于冰浴中12 h。當濾液中晶體不再析出時,抽濾,分離,洗滌,重結晶得到白色結晶,即為產物己二酸。試驗證明該產物的傅里葉變換紅外光譜(FT-IR)和熔點(151~153 ℃)與標準樣品一致[8]。本研究中,己二酸產率的計算式如下:

(1)
1.4 催化劑表征
采用美國Perkin-Elmer公司生產的Spectrum One型紅外光譜儀表征催化劑的結構,具體參數為:分辨率2 cm-1,光譜范圍4 000~400 cm-1。采用日本SHIMADZU公司生產的SSX-550型掃描電子顯微鏡(SEM)觀察催化劑形貌特征,觀察前樣品需在真空、電流50 mA條件下噴金處理。采用日本理學公司生產的D/max-RB型X射線衍射(XRD)儀表征催化劑的晶體結構及組成,Cu靶,Kα射線,管壓40 kV,管流40 mA,掃描速率5(°)/min,掃描范圍5°~50°。采用美國康塔儀器公司生產的ASIQM0002-3型吸附儀表征催化劑的比表面積和孔徑(N2吸附-脫附),測試前催化劑需在80 ℃加熱3 h以消除孔道中吸附的水分,然后在溫度-196 ℃下吸附和脫附N2。
2 結果與討論
2.1 催化劑結構分析


圖1 不同催化劑樣品的紅外光譜a—0%PTA@Cu-MOF-74; b—10%PTA@Cu-MOF-74;c—20%PTA@Cu-MOF-74; d—30%PTA@Cu-MOF-74;e—40%PTA@Cu-MOF-74
與0%PTA@Cu-MOF-74相比,加入PTA后催化劑樣品的紅外光譜中出現了新的特征吸收峰,即波數1 052 cm-1和 956 cm-1處的峰分別歸屬于PTA的中心四面體P—O和W=O,而且隨著PTA含量的增加,上述吸收峰的強度也有一定程度的增強,但是波數750 cm-1處沒有出現吸收峰,可能是因為Cu-MOF-74孔道尺寸的限制效應,導致檢測不到該處的吸收峰。至于波數890 cm-1處出現的特征峰應該與PTA結構中的W—Ob—W(Ob為PTA的Keggin結構中的橋氧基團)[10]的振動和2,5-二羥基對苯二甲酸結構中苯環C—H 單鍵面內外搖擺振動二者有關。但催化劑40%PTA@Cu-MOF-74在波數750 cm-1處出現了特征峰,說明催化劑表面有部分PTA出現。綜合上述分析可見,PTA的Keggin結構沒有被破壞,而且成功負載到Cu-MOF-74骨架結構中。
2.2 催化劑形貌分析
合成的x%PTA@Cu-MOF-74的掃描電鏡照片見圖2。由圖2可見,0%PTA@Cu-MOF-74的微觀形貌為光滑的薄片狀晶體,從端面觀察可以看出呈六方棱柱結構;負載PTA后,當負載量(w)小于40%時,其結構沒有發生明顯的變化,仍能保持良好的晶體結構,也未觀察到PTA顆粒團聚現象[10];但當PTA負載量(w)增加到40%時,催化劑表面變得很粗糙,甚至出現了一些顆粒聚集。這可以進一步證實,本研究中比較適宜的PTA負載量(w)為小于40%,此時催化劑仍能保持良好的Cu-MOF-74原有晶體形貌,且PTA很好地分散在金屬框架結構中。

圖2 不同催化劑樣品的SEM照片
2.3 催化劑結構與組成分析
圖3為x%PTA@Cu-MOF-74的XRD圖譜。由圖3可知,沒有負載PTA時Cu-MOF-74的譜圖在2θ為6.8°和11.8°處出現兩個主要特征衍射峰,這與Abedini等[11]合成的Cu-MOF-74的特征衍射峰一致,其他衍射峰的位置也相吻合,說明本研究成功合成了結晶度高且有序的金屬-有機骨架材料Cu-MOF-74。加入PTA后,XRD譜圖中只有峰強度隨著負載量的增加而有所減弱,其他沒有發生變化。PTA的特征衍射峰位置在2θ為10.7°,21.5°,26.4°,35.9°處[12],而PTA負載量(w)為10%~30%時PTA@Cu-MOF-74的XRD譜圖上沒有這些峰,表明PTA高度分散在Cu-MOF-74孔道中,且催化劑仍能很好地保持骨架結構,不受PTA負載量的影響。但是,當PTA負載量(w)為40%時,XRD譜圖在2θ為10.7°,21.5°,26.4°,35.9°處出現了新的特征峰,說明PTA引入量過大會導致催化劑表面出現PTA團聚現象。通過上述表征結果可以看出,PTA負載量并不是越多越好,基于環保與經濟等方面的考慮,后面的研究主要針對10%PTA@Cu-MOF-74催化劑。

圖3 不同催化劑樣品的XRD圖譜a—0%PTA@Cu-MOF-74; b—10%PTA@Cu-MOF-74;c—20%PTA@Cu-MOF-74; d—30%PTA@Cu-MOF-74;e—40%PTA@Cu-MOF-74
2.4 催化劑表面積、孔徑及孔體積分析
圖4為負載PTA前后催化劑在-196 ℃下的N2吸附-脫附等溫線。根據IUPAC的分類標準,0%PTA@Cu-MOF-74和10%PTA@Cu-MOF-74的吸附-脫附曲線均屬于以微孔為主的Ⅰ型等溫吸附-脫附曲線,說明兩種催化劑均有著均勻的孔道。在相對壓力小于0.1時,隨著相對壓力的增加,N2吸附量增加迅速,主要是因為Cu-MOF-74中含有大量的微孔結構。當相對壓力大于0.1后,吸附等溫線增加較平緩,表明催化劑孔道的吸附逐漸達到飽和。最后曲線出現的向上拖尾是由于晶體顆粒團聚形成結構間隙產生的。負載PTA后催化劑的N2吸附量明顯減少,其原因是PTA占據了MOFs晶體的孔道,使得晶體的比表面積和孔體積減小,吸收容納的N2體積隨之減少。以上結果表明,10%PTA@Cu-MOF-74成功負載了活性物質PTA,這對于改善催化劑的穩定性和催化活性至關重要。
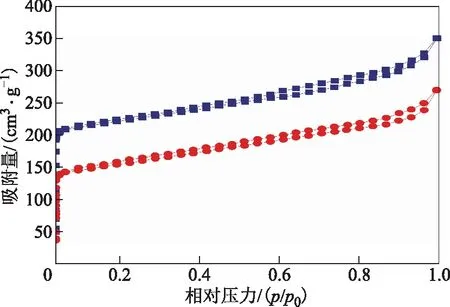
圖4 負載PTA前后催化劑的N2吸附-脫附等溫線■—0%PTA@Cu-MOF-74; ●—10%PTA@Cu-MOF-74
0%PTA@Cu-MOF-74和10%PTA@Cu-MOF-74的BET比表面積、孔體積及孔徑如表1所示。由表1可知,經過高溫活化后的0%PTA@Cu-MOF-74的BET比表面積為1 096 m2/g,平均孔徑為1.01 nm,與文獻[1]的研究結果基本一致,而在負載一定質量PTA后,催化劑的BET比表面積、孔體積及孔徑均有所減小,說明PTA的引入占據了MOFs材料的孔道,也能穩定地負載在金屬有機骨架材料的孔道中。通過上述的分析可以發現,PTA的引入充分利用了MOFs材料穩定的骨架結構,剛性骨架結構能有效減少活性物質的流失,為反應體系提供了一個高效的催化環境,也為反應的進行提供了保障。

表1 催化劑的比表面積、孔體積及孔徑表征結果
2.5 PTA@Cu-MOF-74的催化活性
由H2O2氧化環己烯制己二酸的機理可知,理論上1 mol環己烯氧化生成己二酸需要消耗4 mol H2O2[13],但反應過程中H2O2會有部分分解損失,因此本研究控制H2O2過量10%,即n(環己烯)∶n(H2O2)=1∶4.4[14]。如圖5所示,環己烯氧化成己二酸發生在雙鍵上,大致分6個步驟,其間包括3種氧化反應(烯烴環氧化、醇的氧化和Baeyer-Villiger氧化反應)和2種水解反應(環氧水解和開環水解反應),環己烯在酸性條件下經環氧化后先得到環己二醇,再繼續氧化開環得到己二酸。

圖5 環己烯合成己二酸的反應過程[15]
取1 mL環己烯、4.5 mL 30%H2O2,進行催化反應試驗,考察催化劑用量、反應溫度、反應時間以及催化劑上PTA負載量對PTA@Cu-MOF-74催化H2O2氧化環己烯合成己二酸產率的影響,結果分別如圖6~圖9所示。

圖7 反應溫度對己二酸產率的影響反應時間為6 h,催化劑為10%PTA@Cu-MOF-74,用量為0.2 g

圖8 反應時間對己二酸產率的影響反應溫度為80 ℃,催化劑為10%PTA@Cu-MOF-74,用量為0.2 g

圖9 PTA負載量對己二酸產率的影響反應溫度為80 ℃,反應時間為6 h,催化劑用量為0.2 g
由圖6可以看出,隨著催化劑用量的增加,己二酸的產率先增高而后緩慢降低,在10%PTA@Cu-MOF-74用量為0.2 g時,己二酸產率達到最高,為61.96%。原因可能是主副反應的氧化過程均需要氧化劑的存在,而相對過剩的催化劑活性中心會加速H2O2的分解或促進其他副產物生成;同時,過多的H2O2使環己烯發生了過度氧化導致己二酸產率降低,生成的己二酸也有可能被過量的H2O2過度氧化為戊二酸、丁二酸等副產物[16]。
由圖7可以看出,隨著反應溫度的升高,己二酸產率先增高后降低,在80 ℃達到最大,原因是溫度過高會導致部分H2O2分解,同時過高的反應溫度也可能使副產物增多,影響主反應,導致己二酸產率下降。
由圖8可以看出:當反應時間為4~6 h時,隨著反應時間的延長,己二酸產率迅速增高,在反應時間為6 h時達到頂峰;之后隨著試驗繼續進行,己二酸產率反而下降,可能是反應時間延長后己二酸部分分解所致。
由圖9可以看出:負載PTA的Cu-MOF-74催化劑有著良好的催化活性,當PTA負載量(w)為10%時,己二酸的產率最高;之后隨著PTA負載量(w)繼續增加,己二酸的產率反而下降。這是因為過多的PTA會進一步占據孔道,而且PTA過多會使體系的pH降低,加劇H2O2的分解并降低其濃度,從而降低了己二酸的產率[17]。相比于樊紅超等[8]采用SBA-15分子篩負載PTA合成己二酸,本研究的催化反應所需時間縮短,催化劑的用量也較少,有著很好的經濟適用性。
2.6 催化劑的穩定性
為了評價10%PTA@Cu-MOF-74催化劑的穩定性,在前述初步優化的反應條件(催化劑用量為0.2 g,反應溫度為80 ℃,反應時間為6 h)下對催化劑進行循環使用,每次使用后取出催化劑,分離,無水甲醇洗滌,在烘箱中于60 ℃、0.04 MPa真空度下干燥24 h,然后再次與新鮮的反應物混合,考察其催化活性。
催化劑循環使用次數對己二酸產率的影響見圖10。其中,循環次數為0代表使用的是10%PTA@Cu-MOF-74的新鮮催化劑。

圖10 催化劑循環使用次數對己二酸產率的影響
由圖10可見:第1次循環使用時,己二酸產率為61.46%,比使用新鮮催化劑時的61.96%僅降低了0.50百分點;當催化劑循環使用第4次時,己二酸產率仍然能夠達到59.89%,與使用新鮮催化劑時相比,降低幅度僅為3.3%。這表明制備的10%PTA@Cu-MOF-74催化劑具有良好的穩定性,可重復使用,且試驗所用的活性恢復方法有效減少了活性物質的流失,使催化劑的活性沒有明顯下降。另外需要說明的是,在催化劑循環使用的過程中發現回收催化劑的質量略有降低,這是因為在反應過程中催化劑存在一定程度的磨損流失。
3 結 論
本研究采用一步法原位制備了不同PTA負載量的x%PTA@Cu-MOF-74催化劑,并將其應用于環己烯氧化制己二酸的催化氧化過程中。結果表明,無需添加任何有機溶劑或相轉移劑,就可獲得較高產率的己二酸(61.96%)。在反應溫度為80 ℃、反應時間為6 h、環己烯體積為1 mL、30% H2O2體積為4.5 mL、10%PTA@Cu-MOF-74催化劑添加量為0.2 g的條件下,己二酸的產率最佳,為61.96%。催化劑重復使用第4次時,己二酸的產率仍較高,其活性降低幅度僅為3.3%,催化劑中PTA的流失情況得到了明顯改善。
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
現代企業(2015年9期)2015-02-28 18:56:50
