十八烷@聚甲基丙烯酸甲酯微膠囊相變材料的制備及表征
2022-12-14 07:24:08申晉琛林明桂馬中義賈麗濤李德寶
燃料化學學報 2022年11期
申晉琛 ,林明桂 ,馬中義,* ,賈麗濤,3 ,李德寶,3,*
(1. 中國科學院山西煤炭化學研究所 煤轉化國家重點實驗室, 山西 太原 030001;2. 中國科學院大學, 北京 100049;3. 中國科學院潔凈能源創新研究院, 遼寧 大連 116023)
基于中國資源稟賦和費托合成技術的逐漸成熟,煤制油產業快速發展,但是由于受到原料煤和燃料油價格的影響,其經濟性一直不佳,如何高值化利用煤制油產品,拓展煤制油產品的應用領域,對提高煤制油產業的經濟性至關重要。費托合成產物中含有大量的正構烷烴,其作為相變材料(Phase change materials,PCMs)具有焓值高和穩定性好等優點,廣泛應用于智能調溫應用[1-5]。然而,由于存在易燃與泄露等問題,正構烷烴的直接使用受到了極大程度的制約[6-10]。微膠囊化相變材料(Microencapsulated PCMs,MePCMs)為現存問題的解決開辟了新途徑。在眾多囊壁材料中,無毒無害、來源廣泛且機械強度更高的聚甲基丙烯酸甲酯備受研究人員青睞[3,11,12]。在眾多合成方法中,乳液聚合法由于操作方便、聚合度高常用于制備微膠囊,但其包埋率較低且影響因素繁多,其中,懸浮穩定劑對于改善微膠囊性能發揮著至關重要的作用。通常情況下,單一型乳化劑或分散劑無法滿足制備需求,因此,采用復合型穩定劑提升乳液的穩定性和微膠囊的熱力學性質成為研究重點之一[13-16]。
鑒于以上分析,本研究選用十八烷(Oct)為相變芯材,聚甲基丙烯酸甲酯(PMMA)為壁材,十二烷基硫酸鈉(SDS)為乳化劑,聚乙烯吡咯烷酮(PVP)為分散劑,過硫酸鉀(KPS)為引發劑,季戊四醇四丙烯酸酯(PETRA)為交聯劑,采用乳液聚合法合成Oct@PMMA微膠囊相變材料,通過傅里葉變換紅外、X射線衍射、掃描電子顯微鏡、差示掃描量熱法以及熱重分析法等表征手段對微膠囊的化學和晶體結構、形貌、熱力學性質以及熱穩定性進行表征,考察復合型懸浮穩定劑SDS/PVP和PETRA的添加對微膠囊性能的影響。
1 實驗部分
1.1 微膠囊的制備
微膠囊制備過程如圖1所示。將SDS和一定量PVP溶于100 mL去離子水中,攪拌10 min獲得均勻水相;8 g Oct分散到水相,以1000 r/min速率機械攪拌30 min;緩慢滴加6.5 g MMA和1.5 g PETRA,繼續攪拌至形成穩定O/W乳液;加入0.8 g KPS進行1 h預聚反應,然后繼續反應5 h形成微膠囊懸浮液;待自然冷卻至室溫,以4000 r/min 轉速離心收集微膠囊,并用60 ℃無水乙醇和去離子水各洗滌三次除去未封裝的Oct,置于60 ℃真空烘箱干燥12 h,所得白色粉末即為Oct@PMMA微膠囊。

圖 1 Oct@PMMA微膠囊合成機制示意圖Figure 1 Scheme of formation mechanism for Oct@PMMA MePCMs
其中,SDS與PVP的化學結構如圖2所示,微膠囊具體形成過程如下:首先,SDS在水中發生電離形成帶正電的疏水基團與帶負電的親水基團,疏水非極性基團朝向Oct與之緊密結合,親水極性基團朝向水相,從而在Oct周圍產生強烈的負電子場,同時,PVP分子長鏈在Oct表面充分伸展,使得Oct在SDS的靜電穩定作用與PVP的空間位阻作用下分散成細小液滴[17,18];然后,隨著溶液中MMA濃度降低,增溶膠束中的MMA逐漸解析出來并吸附至Oct液滴表面。最后,水相中的KPS分解形成活性自由基,擴散至液滴表面并引發MMA聚合形成高聚物,包覆Oct形成Oct@PMMA微膠囊相變材料,微膠囊制備配方列于表1。
圖 2 SDS(a)與PVP(b)的化學結構式Figure 2 Chemical structure of SDS (a) and PVP (b)
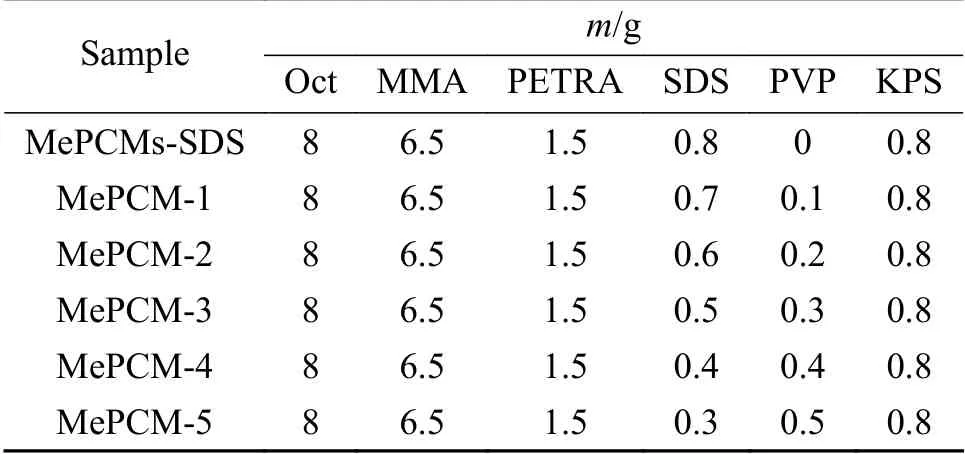
表 1 微膠囊制備配方Table 1 Formulations for the preparation of MePCMs
1.2 微膠囊表征
利用傅里葉變換紅外光譜儀(FT-IR)Nicolet Magna iS10測定微膠囊的化學結構,將干燥的微膠囊與KBr以1∶100的比例混合均勻,取30 mg混合物進行壓片并放入檢測器中采集紅外光譜譜圖,4000-600 cm-1掃描;采用X射線衍射儀(XRD)PANalytical Empyrean X’pert確定微膠囊的晶體結構,以CuKα(λ=0.15406 nm)為輻射源,電壓為50 kV,電流為35 mA,10°-40°掃描,掃描速率為5(°)/min;利用場發射電子顯微鏡(SEM)JSM-7001F對微膠囊的微觀形貌進行觀察并對粒徑進行統計,儀器工作電壓為5 kV;微膠囊的熱重分析(TG)在Setaram TGA-92上進行測試,Ar2氛圍下,以10 ℃/min的升溫速率記錄溫度為40-600 ℃的失重曲線;N2氛圍下,加熱速率為10 ℃/min,測試溫度為0-50 ℃,通過差示掃描量熱儀(DSC)Q200對微膠囊的熱學性能進行測試,得到Oct與微膠囊的熱力學性質,微膠囊的包覆率(Encapsulation ratio,R)、包覆效率(Encapsulation efficiency,E)、熱儲存能力(Thermal storage capability,C)及收率(Yield,Y),計算方法如下。

式中,ΔHm,oct、ΔHm,micro分別代表Oct與微膠囊的熔融焓,ΔHc,oct、ΔHc,micro分別代表Oct與微膠囊的結晶焓,mdried與mtotal分別代表微膠囊干重與原料總質量。
2 結果與討論
2.1 化學結構表征
采用FT-IR對Oct、PMMA和微膠囊的化學組成進行表征,譜圖見圖3。在Oct譜圖中,2915和2845 cm-1處的高強度峰分別歸屬于甲基的對稱伸縮振動和亞甲基的不對稱伸縮振動,同時,1468和1373 cm-1處的吸收峰是由亞甲基和甲基中C-H的彎曲振動引起的,721 cm-1處的弱吸收峰起因于亞甲基的面內搖擺振動。在PMMA譜圖中,2952、1734 cm-1處的吸收峰分別為C-H與C=O的伸縮振動,而1199、1161 cm-1處的雙吸收峰歸屬于C-O的伸縮振動[19-22]。對比發現,所有微膠囊樣品紅外譜圖中包含Oct與PMMA全部的特征吸收峰且沒有產生新的吸收峰,表明PMMA成功包覆Oct形成微膠囊且兩者之間沒有發生化學反應。
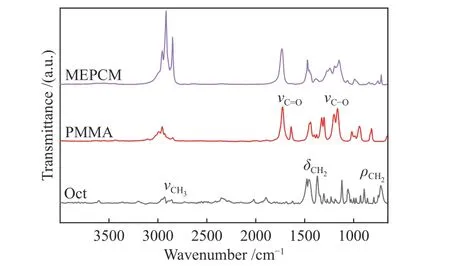
圖 3 十八烷、聚甲基丙烯酸甲酯與微膠囊的FT-IR譜圖Figure 3 FT-IR spectra of Oct, PMMA and MEPCMs
2.2 晶體結構表征
采用X射線粉末衍射對Oct、微膠囊進行晶體結構表征,如譜圖4所示。Oct與微膠囊化的Oct都為三斜晶系且擁有明顯的結晶結構,2θ值為19.24°、23.31°、24.71°與24.84°處的高強度衍射峰分別對應于β相Oct的(010)、(105)、(-101)和(110)面,同時,2θ值為11.59°、15.45°和39.67°處的衍射峰分別對應α相Oct的(003)、(004)和(0-22)面。值得注意的是,微膠囊化的Oct衍射峰強度顯著降低,11.59°、15.45°和24.84°處的衍射峰強度減弱甚至消失,主要原因在微膠囊狹小的空間限制了Oct分子長鏈的運動,導致其結晶度降低,進一步說明微膠囊相變材料的形成[12,19-21]。
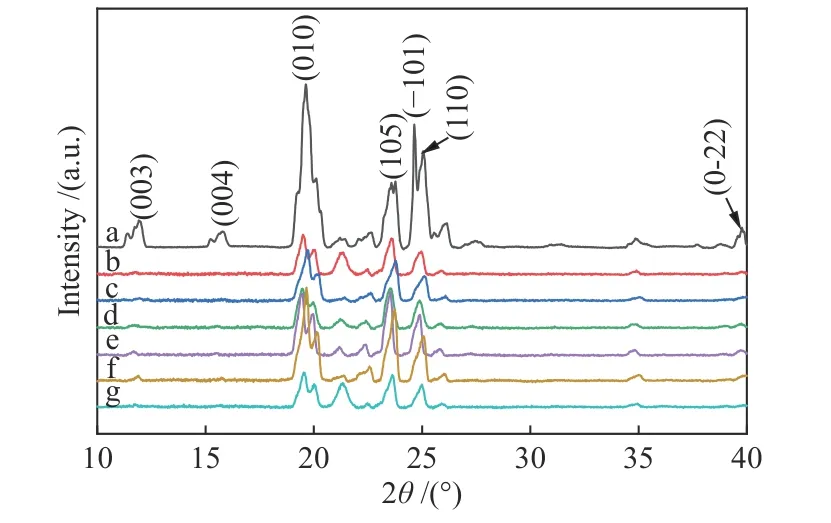
圖 4 Oct(a)與微膠囊(b-g)的XRD譜圖Figure 4 XRD patterns of Oct (a) and MePCMs (b-g)
FT-IR和XRD結果表明,十八烷成功包覆于聚甲基丙烯酸甲酯形成Oct@PMMA微膠囊相變材料。
2.3 微觀形貌表征
采用掃描電子顯微鏡對微膠囊的微觀形貌進行觀察,其SEM照片如圖5所示。由圖5可以看出,微膠囊顆粒大致為球形,粒徑位于10-40 μm,隨著PVP添加量的增加,微膠囊顆粒逐漸發生破裂(c)甚至坍塌(d),表面變得粗糙((f)、(g))且有凹陷(h)產生。可能的原因在于PVP屬于高分子型懸浮穩定劑,在水中難以發生電離,當在油滴表面吸附至飽和之后,使得溶液中開始存在大量PVP分子。隨著聚合反應的進行,單體MMA逐漸生長成為長鏈聚合物,容易與PVP分子長鏈發生相互糾纏,從而延緩了單體向Oct的擴散與吸附,增加了單體在溶液中發生聚合的概率,使得單體未全部參與形成微膠囊殼層結構而生成大量聚合物碎屑((f)、(g)、(h)),堆積至Oct液滴表面對其造成擠壓,導致微膠囊表面粗糙不平且有凹陷產生;同時,單體利用率的大幅降低使得微膠囊殼層厚度變薄,機械強度降低,容易在外力作用下發生破裂,造成相變芯材的泄漏。

圖 5 微膠囊的SEM照片Figure 5 SEM micrographs of MePCMs
2.4 粒徑分布
微膠囊粒徑分布如圖6所示,隨著PVP添加量的增加,微膠囊的平均粒徑及分布范圍先變小后變大,相較于微膠囊整體而言,殼層厚度較薄,因此,乳液液滴粒徑分布呈現出相同的變化規律。均勻穩定的乳液是制備性能優良微膠囊的前提,而乳液的穩定性與液滴保護膜的強度息息相關[14,15]。從微膠囊形成過程角度出發,由于具有較大的空間位阻,PVP的添加進一步提高了液滴保護膜的強度,有效防止了液滴發生聚并,使得Oct與MMA的分散度與均一性增加,液滴粒徑減小,比表面積增大,從而促進了MMA向Oct表面的遷移與吸附,有利于高包覆率的形成。然而,隨著PVP的繼續添加,乳液體系黏度增大使得油相分散難度上升,導致液滴粒徑增大,均一性與穩定性降低,不利于微膠囊相變材料的生成。其中,當PVP分子濃度過大之后,微膠囊粒徑呈現多分散分布,可能是由于生成大量聚合物碎屑引起的,與2.3中觀測到的現象一致。
2.5 熱力學性質表征
作為微膠囊最重要的熱力學性能參數,包覆率描述了微膠囊對于相變材料的有效封裝,包覆效率代表了微膠囊內相變材料的有效表現。利用差示掃描量熱法對芯材與微膠囊的熱力學性質進行測試,相變溫度和相變焓列于表2。熔融曲線與結晶曲線如圖7(a)與7(b)所示,隨著PVP用量的增加,微膠囊吸熱峰向低溫方向遷移,放熱峰向高溫方向遷移,可能的原因是微膠囊殼層厚度變薄導致內外熱交換速率加快,與2.3中形貌觀察所得結論一致。
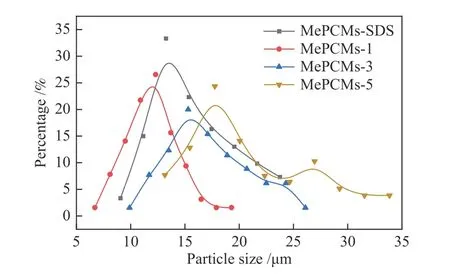
圖 6 微膠囊粒徑分布Figure 6 Particle size distribution of MePCMs
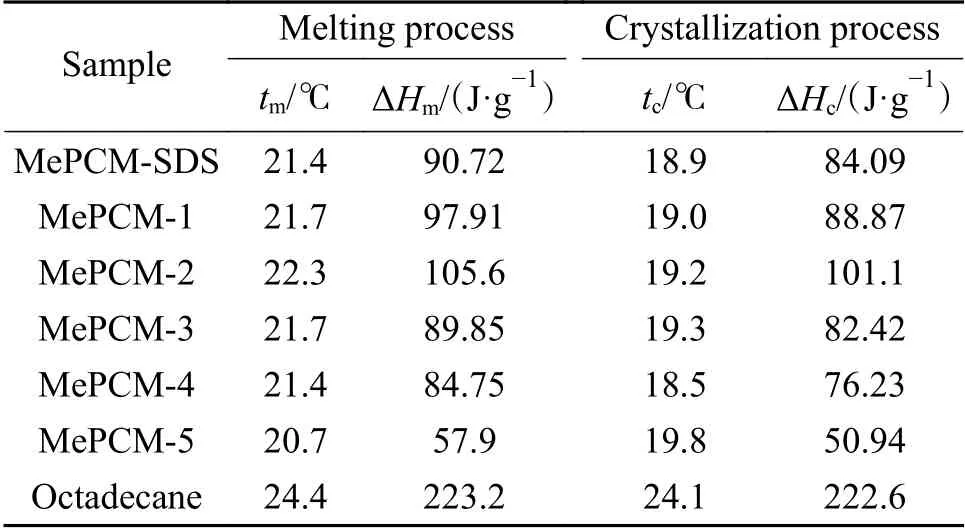
表 2 微膠囊熱力學性質Table 2 Thermal properties of MePCMs
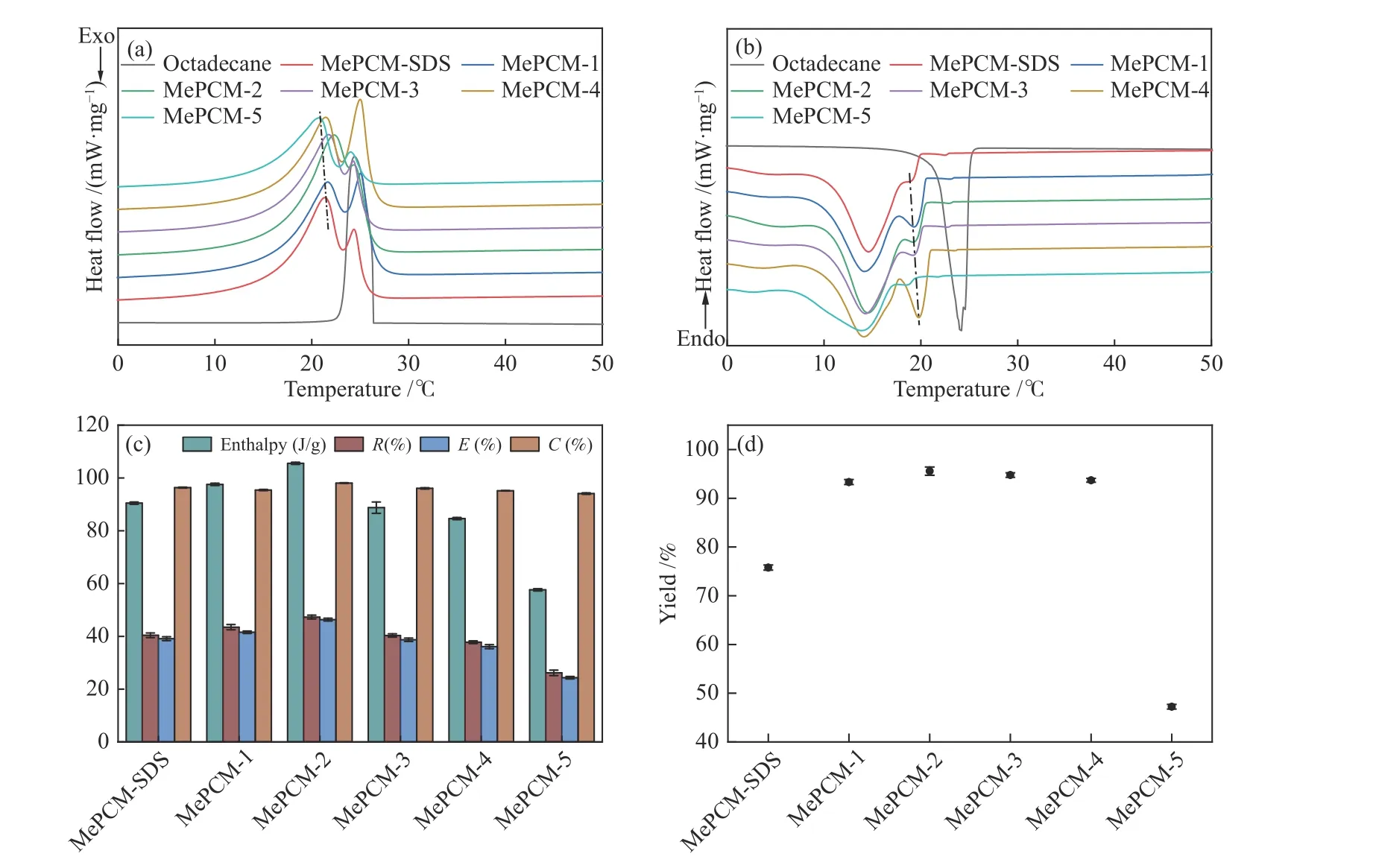
圖 7 (a):DSC熔融曲線;(b):DSC結晶曲線;(c):相變焓值、包覆率、包覆效率和熱儲存能力;(d):收率Figure 7 (a): DSC Heating curves; (b): DSC cooling curves; (c): phase change enthalpies, R, E and C; (d): Y
由圖7(c)可知,微膠囊相變焓值呈現先上升后下降的趨勢,焓值最高為105.6 J/g,包覆率呈現出相同的變化規律,說明焓值大小依賴于微膠囊對Oct的有效封裝;同時,包覆率與包覆效率之間相差較小,熱儲存能力均高于95%,表明膠囊化的Oct基本全部可以發生相變進行潛熱儲存;此外,當PVP添加量為0.2 g時,微膠囊包覆率最高為47.31%,收率高達98.01%,原因在于PVP分子在液滴表面的吸附達到飽和且溶液中的分散劑濃度較低,最大限度地提升了保護膜的強度,使得乳液的穩定性與液滴的分散度得到顯著改善,從而促進MMA向Oct的遷移,提升了微膠囊的包覆率與產率。
2.6 熱穩定性表征
采用熱重分析法對Oct和微膠囊的熱降解行為進行表征,失重曲線如圖8所示。Oct經過一步分解后完全失重,起始分解溫度為110.5 ℃,且在280 ℃分解完全。所有微膠囊樣品具有相似的兩步失重過程,第一階段失重溫度為150-280 ℃,歸因于Oct的擴散與分解,失重率與DSC結果基本一致;第二階段失重溫度為300-500 ℃,主要是由PMMA分解引起的。對于第一階段,Oct的起始分解溫度由110.5 ℃上升至150 ℃,證明微膠囊化可以有效防止Oct泄露并提高其分解溫度[23-25]。
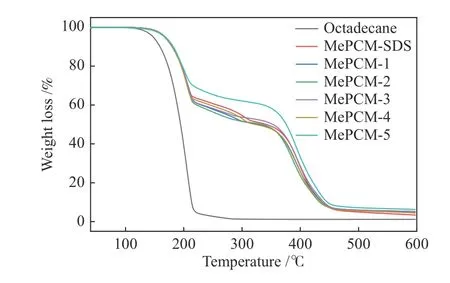
圖 8 Oct與微膠囊熱重曲線Figure 8 TG curves of Oct and MePCMs
在保持殼層材料總量不變的前提下,通過調變MMA與PETRA的比例,考察其對微膠囊熱穩定性的影響。由圖9(a)知,當MMA/PETRA分別為7∶1、6.5∶1.5、6∶2時,微膠囊第一階段失重曲線基本相似,說明微膠囊中Oct的質量分數大致相同;隨著PETRA添加量增加,微膠囊分解溫度提升至175 ℃,原因在于線型MMA分子之間通過PETRA的交聯作用形成化學鍵,使其形成三維網絡空間結構,MMA之間化學鍵合作用增強。當MMA/PETRA為5.5∶2.5時,盡管分解溫度進一步提升,但微膠囊中Oct質量分數急劇下降,原因在于過量PETRA導致MMA交聯過快,PMMA韌性變差,微膠囊容易發生破損,造成封裝的Oct在離心、洗滌等過程中被除去。此外,微膠囊MePCM-6-2相變循環曲線與對應的焓值如圖9(b)與9(c)所示,發現經過100次相變循環之后,微膠囊相變趨勢基本保持不變,相變焓值下降幅度僅為1.4%。因此,當MMA/PETRA為3∶1時,微膠囊的熱穩定性最好。
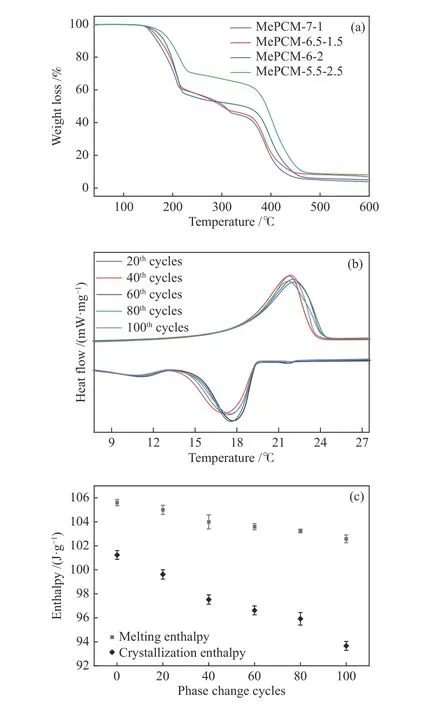
圖 9 (a):不同MMA:PETRA微膠囊熱重曲線;(b):MePCM-6-2的相變循環曲線;(c):MePCM-6-2的相變焓值Figure 9 (a): TG curves of MePCMs at different mass ratio of MMA to PETRA; (b): Phase change cycle of MePCM-6-2;(c): Phase change enthalpy of MePCM-6-2
3 結 論
通過乳液聚合法合成了Oct@PMMA微膠囊,使用SDS與PVP作為復合型懸浮穩定劑,PETRA作為交聯劑,得到相變潛熱較高、收率較高以及熱穩定性較好的微膠囊。結果表明,SDS的靜電穩定和PVP巨大的空間位阻作用增強了乳液液滴的穩定性與分散程度,提升了微膠囊的包覆率和收率;此外,PETRA的增加進一步提升了微膠囊的熱穩定性。Oct@PMMA是一種具有廣闊應用前景的相變儲熱材料,進一步拓展了費托合成產物的應用領域。
