UPLC指紋圖譜結合化學計量學方法比較當歸和歐當歸藥材的成分差異
2022-11-18 02:25:38王亞丹陳明慧閆建功姚令文馬雙成
中草藥 2022年22期
關鍵詞:差異
王亞丹,陳明慧,閆建功, 2,馬 蕭,姚令文*,戴 忠*,馬雙成
UPLC指紋圖譜結合化學計量學方法比較當歸和歐當歸藥材的成分差異
王亞丹1,陳明慧1,閆建功1, 2,馬 蕭3,姚令文1*,戴 忠1*,馬雙成1
1. 中國食品藥品檢定研究院,北京 100050 2. 河北中醫學院 河北省中藥炮制技術創新中心,河北 石家莊 050200 3. 甘肅省藥品檢驗研究院,甘肅 蘭州 730070
建立當歸和歐當歸藥材的指紋圖譜,結合化學計量學方法尋找二者的成分差異,從而為建立歐當歸摻偽當歸的鑒別方法提供依據。采用UPLC法建立指紋圖譜,色譜柱為Waters Cortecs T3柱(100 mm×2.1 mm,1.6 μm);流動相為0.1%甲酸乙腈溶液-0.1%甲酸水溶液,梯度洗脫;體積流量為0.4 mL/min;柱溫35 ℃;檢測波長為210 nm。采用相似度評價、主成分分析(principal component analysis,PCA)和層次聚類分析(hierarchical cluster analysis,HCA)對2種藥材的指紋圖譜進行評價,尋找2者的成分差異。采用UPLC-MS/MS及NMR波譜等技術對主要差異性成分進行鑒定。從當歸和歐當歸指紋圖譜中分別識別出23個和19個共有峰,與當歸指紋圖譜共有模式相比,當歸樣品組內有3批樣品相似度較低,為0.59~0.80,其余樣品相似度均大于0.99,而歐當歸與之相比,相似度在0.53~0.94,且組內差異較大。PCA和HCA結果顯示,當歸和歐當歸可明顯分為2組,對區分貢獻較大的色譜峰有6個,鑒定為色氨酸、阿魏酸、-藁本內酯、歐當歸內酯A異構體、綠原酸和法卡林二醇,其中后2種成分在歐當歸中含量較高,其余成分在當歸中含量較高。建立的UPLC指紋圖譜結合化學計量學評價方法,簡便可行,可有效區分當歸和歐當歸并揭示其差異性成分,為當歸的質量控制及摻偽歐當歸鑒別方法建立提供依據。同時首次從歐當歸中分離得到法卡林二醇,該化合物為歐當歸區別于當歸的主要成分。
當歸;歐當歸;UPLC指紋圖譜;化學計量學;法卡林二醇;色氨酸;阿魏酸;-藁本內酯;歐當歸內酯A異構體;綠原酸
當歸為傘形科當歸屬植物當歸(Oliv.) Diels.的干燥根,主產于我國甘肅、陜西、云南等地,具有補血活血、調經止痛、潤腸通便的功效[1],廣泛用于中醫臨床各科,素有“婦科人參”“十方九歸”之譽[2]。1957年,由于當歸需求量大,資源緊缺,我國從保加利亞引種了同科不同屬植物歐當歸Koch.,在華北、甘肅等地大量栽培,將其根作為當歸的代用品[3]。但在后期臨床使用中發現歐當歸容易引起患者惡心、頭昏或血熱妄行等不良反應,與當歸的溫補作用相去甚遠,因此在20世紀80年代,國家衛生部藥政部門規定不能以歐當歸代替當歸使用[4-5]。然而,由于歐當歸易于栽培,生長期短,目前仍有部分地區栽培,制成飲片后作為當歸或混入當歸中使用,給臨床用藥帶來風險,因此有必要從多方面對2種藥材進行比較并加以區分,建立摻偽鑒別方法。目前對當歸和歐當歸的比較研究多數集中在性狀和顯微特征上[6-8],而在化學成分方面,缺乏對歐當歸的系統研究,僅少數文獻報道當歸和歐當歸二者成分類似[9-10],均含有有機酸、丁苯酞類、氨基酸等成分,但成分差異并不清楚。基于以上情況,本實驗建立了UPLC指紋圖譜結合相似度評價、主成分分析(principal component analysis,PCA)和聚類分析(hierarchical cluster analysis,HCA)等化學計量學方法比較了當歸和歐當歸的成分差異,并采用液質聯用、提取分離及NMR波譜等方法對差異性成分進行結構鑒定,從而為歐當歸摻偽當歸的鑒別方法建立提供依據。
1 儀器與試藥
1.1 儀器
Waters Acquity UPLC色譜系統(包含二元超高壓泵系統、樣品管理系統、PDA檢測器、柱溫箱、Empower色譜工作站)、Waters Synapt G2-S Q-TOF MS液質聯用系統(包括ESI離子源、Masslynx軟件)、Waters全自動純化系統、Waters e2695高效液相色譜系統(美國Waters公司);Bruker AVANCE NEO 600型核磁共振波譜儀(瑞士Bruker公司);AE-240型電子分析天平(瑞士Mettler公司);KQ-300DA型數控超聲波清洗器(昆山市超聲儀器有限公司);Milli-Q純水儀(美國Millipore公司);ChemPattern化學計量學軟件(科邁恩科技有限公司)。
1.2 試藥
對照品藁本內酯(批號111737-201608,質量分數大于95%)購自中國食品藥品檢定研究院;甲醇、乙腈、甲酸為色譜純購自美國Thermo Fisher公司;其他溶劑及硅膠(100~200目)為分析純,購自國藥集團化學試劑有限公司;水為純化水。
實驗所用藥材經中國食品藥品檢定研究院康帥副研究員鑒定13批當歸藥材(編號S1~S13)為傘形科植物.(Oliv.) Diels.的根,13批歐當歸藥材(編號C1~C13)為傘形科植物歐當歸.Koch.的干燥根,具體信息見表1。
2 方法與結果
2.1 對照品溶液的制備
取藁本內酯對照品適量,精密稱定,加甲醇制成質量濃度為0.17 mg/mL的溶液,即得。

表1 當歸和歐當歸樣品來源信息
2.2 供試品溶液的制備
將當歸及歐當歸藥材粉碎,過40目篩,取樣品約0.5 g,精密稱定,置于50 mL具塞錐形瓶中,精密加入70%甲醇20 mL,稱定質量,超聲(300 W、45 kHz)提取45 min,放冷,再次稱定質量,用甲醇補足減失的質量,濾過,取續濾液過0.45 μm濾膜,即得。
2.3 色譜條件
色譜柱:Waters Cortecs T3柱(100 mm×2.1 mm,1.6 μm);柱溫35 ℃;流動相為0.1%甲酸乙腈溶液(A)-0.1%甲酸水溶液(B),梯度洗脫:0~6.0 min,10%~34% A;6.0~6.5 min,34%~40% A;6.5~10.0 min,40% A;10.0~13.0 min,40%~57% A;13.0~17.5 min,57% A;17.5~18.5 min,57%~72% A;18.5~25.0 min,72%~95% A;體積流量0.4 mL/min;檢測波長210 nm;進樣體積3 μL。在上述條件下得到的對照品及代表性樣品色譜圖見圖1。
2.4 質譜條件
離子源:電噴霧離子源(ESI);掃描方式:正、負離子掃描;掃描模式:MSE模式;毛細管電壓:正離子3.0 kV,負離子2.5 kV;錐孔電壓40 V;脫溶劑溫度:450 ℃;脫溶劑(N2)體積流量:1000 L/h;掃描范圍:50~1500;掃描時間0.2 s;碰撞氣體為高純氬氣。設定高低2個能量通道,低能量通道碰撞能量為8 V,高能量通道碰撞能量為20~30 V的梯度能量。
2.5 方法學考察
2.5.1 精密度試驗 取當歸樣品(S12),按“2.2”項下方法制備供試品溶液,按“2.3”項下色譜方法連續進樣6次,以藁本內酯為參照峰,計算其他各特征峰的相對保留時間和相對峰面積,結果各特征峰的相對保留時間RSD值小于0.053%,相對峰面積的RSD值小于1.31%,表明儀器精密度良好。
2.5.2 穩定性試驗 取“2.2”項下供試品溶液(S12),分別于0、2、4、8、12、24 h按“2.3”項下色譜條件進行測定,以藁本內酯為參照峰,計算圖譜中各特征峰的相對保留時間和相對峰面積,結果RSD值分別小于0.30%和2.50%。
2.5.3 重復性試驗 取同一批當歸樣品(S12)6份,按“2.2”項下方法制備供試品溶液并按“2.3”項下色譜條件測定,結果圖譜中各特征峰的相對保留時間和相對峰面積的RSD值分別小于0.24%和2.35%。
2.6 指紋圖譜的建立
取13批當歸和13批歐當歸樣品,分別按照“2.2”項下方法制備供試品溶液,并按“2.3”項下色譜條件測定,將采集的色譜圖導入ChemPattern軟件,分別得到2種藥材的疊加圖譜,見圖2,以50%以上樣品中存在的色譜峰作為共有峰篩選條件,利用高斯曲線模擬的方法生成共有模式,見圖3。在當歸樣品中共識別出23個共有峰,而在歐當歸樣品中共識別出19個共有峰。
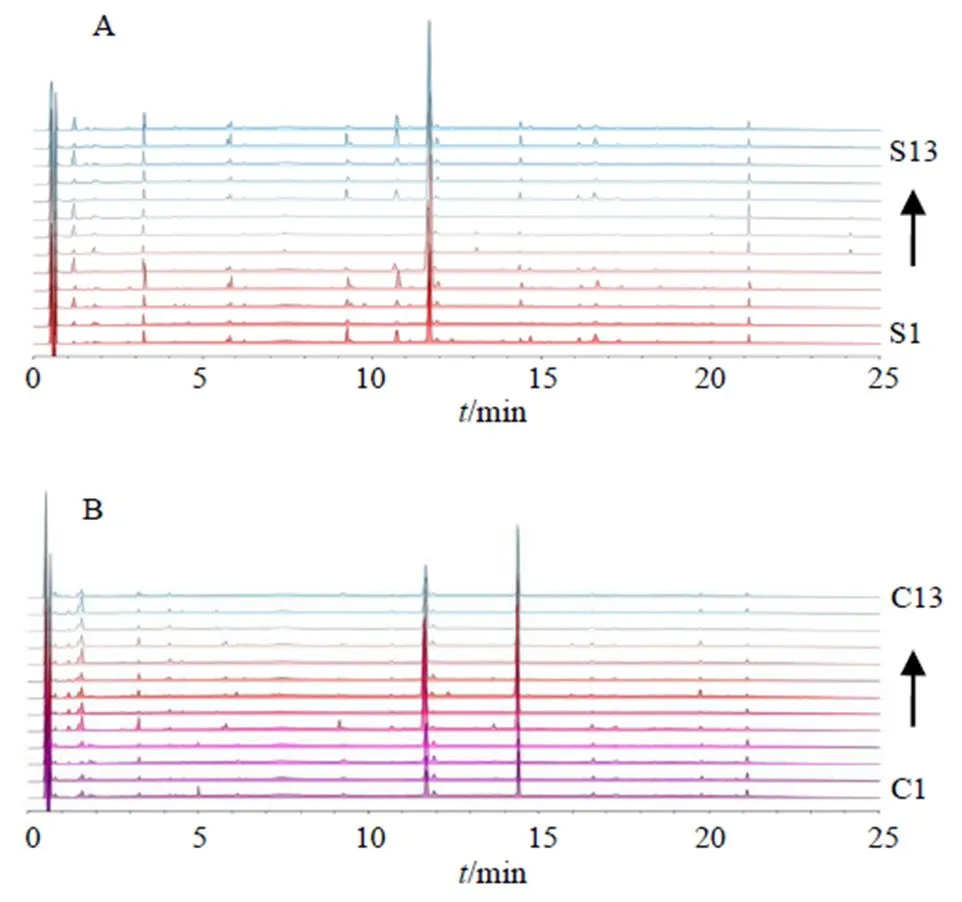
圖2 當歸(A)和歐當歸(B)樣品的UPLC指紋圖譜
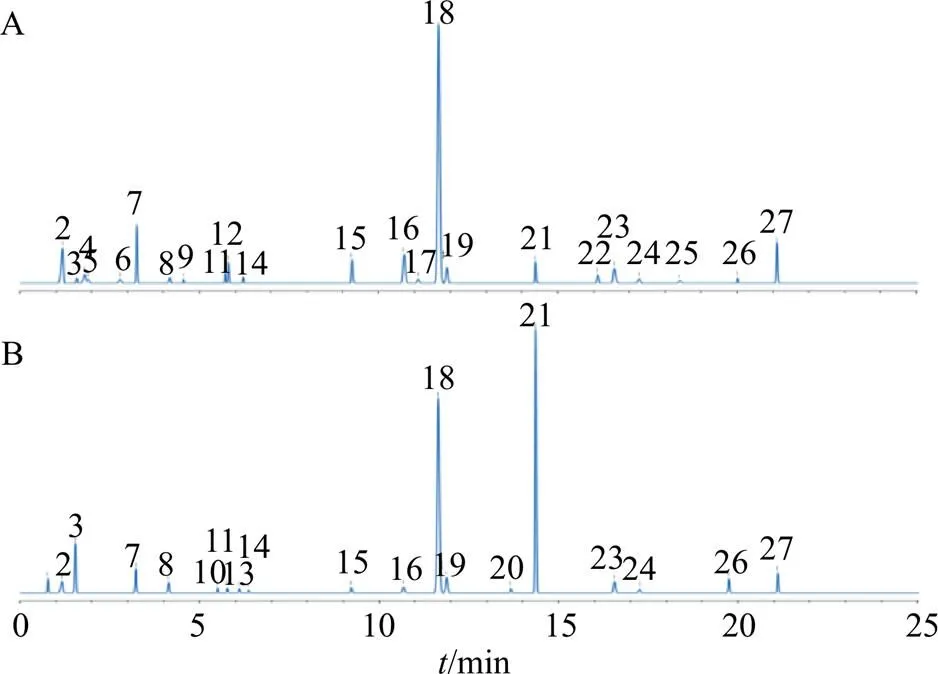
圖3 當歸(A)和歐當歸(B)的UPLC指紋圖譜共有模式
2.7 統計學分析
2.7.1 相似度分析 利用ChemPattern軟件,以當歸UPLC指紋圖譜共有模式為參照圖譜,采用夾角余弦法計算各樣品的相似度,結果見圖4。13批當歸中,除S6~S8外,其余樣品組內相似度良好,在0.99以上;而歐當歸與當歸相比,相似度在0.53~0.94,組內差異較大。
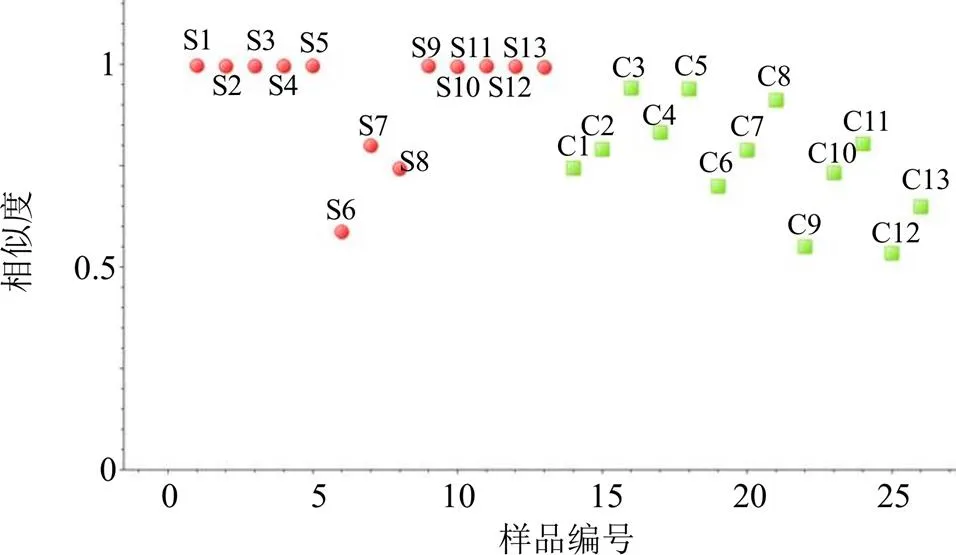
圖4 當歸(S1~S13)和歐當歸(C1~C13)樣品的相似度分析結果
2.7.2 PCA 采用ChemPattern軟件,以當歸和歐當歸UPLC指紋圖譜中共有峰面積為變量,進行PCA,結果見圖5。共提取2個主成分,累積方差貢獻率達到92.1%,說明PCA能較全面地反映樣品差異。得分圖顯示,當歸和歐當歸樣品可以沿PC1軸分別聚為一類;載荷圖顯示,峰2、3、7、18、21和27對PC1的貢獻較大,是當歸和歐當歸中的主要差異性成分,其中峰2、7、18和27在當歸中含量較高,而峰3和峰21則在歐當歸樣品中含量較高。
2.7.3 HCA 以當歸和歐當歸UPLC指紋圖譜中共有峰面積為變量,將樣品圖譜標準化后,以街區距離為度量,采用誤差平方和法進行HCA,結果見圖6。與PCA結果類似,當歸和歐當歸樣品可分別聚成一類,而在當歸樣品中S6~S8與其他批次差異較大。
2.8 主要差異性成分的鑒定
按照“2.3”項下的色譜條件及“2.4”項下的質譜條件對樣品溶液進行分析,對主要差異性成分(峰2、3、7、18、21和27)進行鑒定,在低碰撞能量通道下得到各成分的分子離子峰,高碰撞能量通道下得到成分的碎片信息,結合各成分的色譜行為(圖3),與自建的數據庫進行比較,鑒定結果見表2。其中2、3、7和18號峰采用對照品比對的方式進行確證,而27號峰與歐當歸內酯A的分子式和碎片離子均相同,但保留時間有差別,因此推測其可能為歐當歸內酯A的順反異構體。另外21號峰在正、負離子模式下的質譜峰均較為雜亂,無法推測其分子式,但由于該成分是當歸和歐當歸的主要差異成分,因此有必要對其進行分離純化,得到目標單體后,采用NMR等其他技術進行結構鑒定。

圖5 當歸和歐當歸樣品主成分分析得分圖(A)和載荷圖(B)
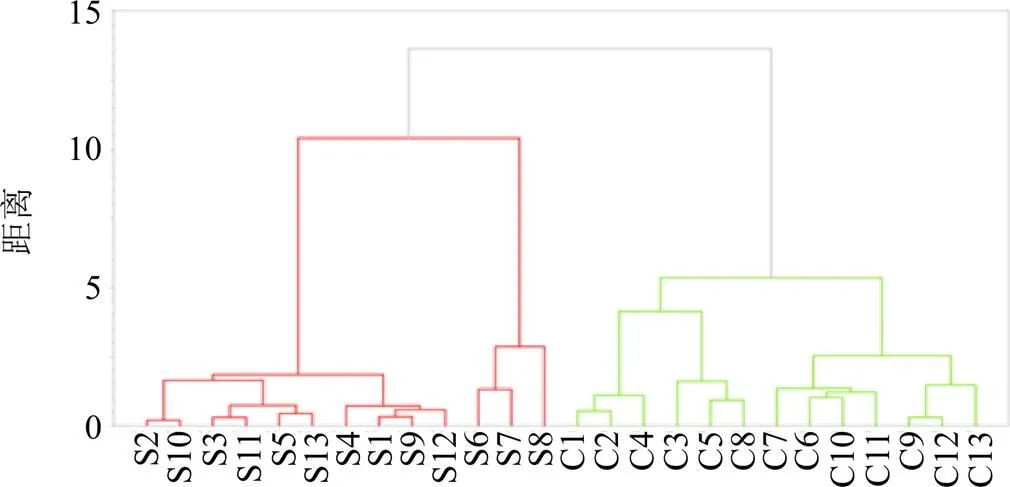
圖6 當歸(S1~S13)和歐當歸(C1~C13)樣品的聚類分析結果
峰21的分離鑒定:取干燥的歐當歸藥材(C1)500 g,粉碎成粗粉,加入1 L 70%甲醇溶液,浸泡12 h,超聲(300 W,45 kHz)提取3次,每次1 h,合并提取液,減壓濃縮至無甲醇。濃縮液依次用石油醚(60~90 ℃)和醋酸乙酯萃取3次,每次500 mL,減壓濃縮后得石油醚部位3.12 g、醋酸乙酯部位6.17 g。將醋酸乙酯部位進行硅膠柱色譜分離,以石油醚(60~90 ℃)-醋酸乙酯(20∶1~3∶1)為流動相,梯度洗脫,經HPLC檢測,合并含目標物的組分,減壓蒸干溶劑后得490 mg。該組分使用Waters X-Bridge Prep OBD C18制備色譜柱(150 mm×30 mm,10 μm),經Waters全自動純化系統進行分離純化,以乙腈-水(60∶40)為流動相,體積流量20 mL/min,檢測波長210 nm,制備得到目標成分107 mg。

表2 當歸與歐當歸中差異性成分的鑒定結果
目標成分為無色油狀物,UV(MeOH)λmax:199, 234, 243, 257 nm。1H-NMR (600 MHz, CDCl3): 5.94 (1H, ddd,= 17.0, 10.2, 5.4 Hz, H-2), 5.61 (1H, dt,= 10.2, 7.2 Hz, H-10), 5.49 (1H, m, H-9), 5.47 (1H, d,= 16.8 Hz, H-1a), 5.26 (1H, d,= 10.2 Hz, H-1b), 5.20 (1H, d,= 8.4 Hz, H-8), 4.94 (1H, brd,= 4.8 Hz, H-3), 2.11 (2H, q,= 7.2 Hz, H-11), 1.39 (2H, m, H-12), 1.22~1.34 (8H, m, H-13~15, 16), 0.88 (3H, t,= 6.6 Hz, H-17);13C-NMR (150 MHz, CDCl3): 117.4 (C-1), 135.8 (C-2), 63.5 (C-3), 79.9 (C-4), 70.3 (C-5), 68.7 (C-6), 78.2 (C-7), 58.6 (C-8), 127.6 (C-9), 134.7 (C-10), 31.8 (C-11), 29.3 (C-12), 29.2 (C-13), 29.1 (C-14), 27.7 (C-15), 22.6 (C-16), 14.1 (C-17)。以上數據與文獻報道一致[11-12],故鑒定該成分為法卡林二醇,其結構見圖7,該化合物首次從歐當歸中分離得到。
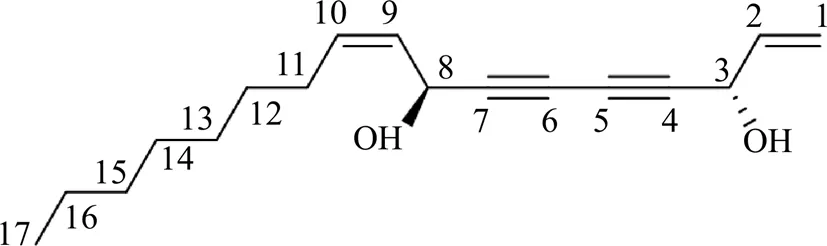
圖7 法卡林二醇(峰21)的結構
3 討論
3.1 供試品前處理方法考察
本研究考察了不同提取溶劑(50%、70%、100%甲醇)、提取方式(超聲和回流)以及提取時間(30、45和60 min)對色譜圖的影響。結果發現,70%甲醇提取的供試品溶液色譜峰數量最多,峰面積中等,而甲醇提取的供試品溶液雖然個別色譜峰面積略大,但由于溶劑效應導致峰形較差,因此選擇70%甲醇作為提取溶劑。在提取方式上,超聲和回流提取效率接近,但前者更方便,另外提取時間超過45 min后對色譜峰面積影響不大,因此,將70%甲醇超聲提取45 min作為最終供試品前處理方法。
3.2 色譜條件的選擇和優化
本研究采用PDA檢測器對當歸和歐當歸樣品溶液進行200~400 nm全波長掃描,發現許多成分的紫外吸收特征存在差異,因此在不同波長下得到的色譜圖中峰數量和面積有明顯區別,考慮到多數成分在低波長210 nm下有吸收,且該波長下2種藥材的色譜圖差異較大,故選擇210 nm作為檢測波長。流動相選擇上,考察了乙腈-水和甲醇-水系統,發現乙腈的分離效果優于甲醇,由于樣品中含有許多有機酸類成分,容易拖尾,因此在流動相系統中加入甲酸,考慮到低波長下色譜圖基線隨流動相pH變化明顯,最終在乙腈和水中均加入0.1%甲酸,使流動相pH相對穩定,基線較為平坦。
3.3 當歸和歐當歸的化學成分比較
本研究采用UPLC指紋圖譜對當歸和歐當歸的化學成分進行整體表征,并結合相似度分析、PCA及HCA等化學計量學方法尋找二者的成分差異,結果發現2種藥材所含成分類型相似,并無專屬性成分,但各成分含量比例上存在較明顯差異。經液質聯用及NMR等技術對差異貢獻較大的成分鑒定后得出:當歸中色氨酸、阿魏酸、藁本內酯以及歐當歸內酯A異構體的含量較高,而歐當歸中綠原酸和法卡林二醇的含量較高,尤其是法卡林二醇,是歐當歸區別于當歸的最主要成分,同時首次在該植物中分離得到。經文獻查閱,法卡林二醇主要存在于傘形科植物中[13-15],具有顯著的抗菌、抗腫瘤和抗炎活性[16-18],推測其可能是造成歐當歸藥性較當歸強烈的原因之一。由于價格、產量等原因,目前歐當歸摻偽當歸的情況時有發生,給臨床用藥帶來較大風險,根據本研究結果,提出2種解決方案,一種是以法卡林二醇作為指標成分,通過控制其含量限度來建立歐當歸摻偽當歸的鑒別方法,另一種是再從上述差異性成分中選擇1個當歸中含量較高的成分,如阿魏酸,以法卡林二醇與該成分的含量比值作為指標,通過控制該比值的限度來建立摻偽鑒別方法。
利益沖突 所有作者均聲明不存在利益沖突
[1] 中國藥典 [S]. 一部. 2020: 139.
[2] 王華, 孫娜. 當歸的有效化學成分及藥理作用研究進展分析 [J]. 山東化工, 2017, 46(18): 59-60.
[3] 王翰華, 胡雙豐. 歐當歸的研究進展 [J]. 中國醫藥導報, 2011, 8(15): 11-12.
[4] 胡雙豐. 歐當歸不可代替中藥當歸藥用 [J]. 中國當代醫藥, 2009, 16(4): 41.
[5] 運泓, 張治國. 當歸與歐當歸的鑒別 [J]. 中藥材科技, 1981, 4(2): 32.
[6] 傅紅. 當歸、歐當歸和日本當歸的鑒別比較研究 [J]. 天津藥學, 2019, 31(6): 15-19.
[7] 溫子帥, 劉愛朋, 景松松, 等. 當歸和歐當歸的定性與定量鑒別 [J]. 中成藥, 2018, 40(12): 2719-2723.
[8] 肖吉元, 楊扶德, 羅文蓉, 等. 當歸與歐當歸的鑒定學比較研究 [J]. 西部中醫藥, 2012, 25(8): 32-35.
[9] 方洪鉅, 呂瑞綿, 劉國聲, 等. 揮發油成分的研究: II.中國當歸與歐當歸主要成分的比較 [J]. 藥學學報, 1979, 14(10): 617-623.
[10] 樂巍, 吳玉蘭, 邱蓉麗. 基于HPLC及化學計量法對當歸與歐當歸藥材的比較研究 [J]. 中藥材, 2018, 41(8): 1846-1850.
[11] Dang N H, Zhang X F, Zheng M S,. Inhibitory constituents against cyclooxygenases fromThunb. [J]., 2005, 28(1): 28-33.
[12] Yamazoe S, Hasegawa K, Suenaga K,. Growth inhibitory polyacetylenes from galls ofbean [J]., 2006, 1(2): 87-94.
[13] 楊秀偉, 嚴仲鎧, 顧哲明, 等. 寬葉羌活化學成分的研究 [J]. 中國藥學雜志, 1994, 29(3): 143.
[14] 林立五, 時嘉敏, 王建忠, 等. 羌活的化學成分研究 [J]. 華西藥學雜志, 2020, 35(1): 28-31.
[15] 石明睿, 趙建喜, 馮祖飛, 等. 迷果芹根化學成分的研究 [J]. 天然產物研究與開發, 2012, 24(2): 182-184.
[16] Zhao C Y, Zheng H D, Zhou L M,. Falcarindiol isolated frominhibits the quorum sensing of[J]., 2021, 26(19): 5896.
[17] Wang C Z, Luo Y, Huang W H,. Falcarindiol and dichloromethane fraction are bioactive components in: Colorectal cancer chemoprevention via induction of apoptosis and G2/M cell cycle arrest mediated by cyclin A upregulation [J]., 2021, 19(2): 113-124.
[18] Venkatensan T, Choi Y, Lee J,. Falcarindiol inhibits LPS-induced inflammation via attenuating MAPK and JAK-STAT signaling pathways in murine macrophage RAW 264.7 cells [J]., 2018, 445 (1/2): 169-178.
Comparison of chemical constituents betweenandby UPLC fingerprints combined with chemometrics methods
WANG Ya-dan1, CHEN Ming-hui1, YAN Jian-gong1, 2, MA Xiao3, YAO Ling-wen1, DAI Zhong1, MA Shuang-cheng1
1. National Institutes for Food and Drug Control, Beijing 100050, China 2. Traditional Chinese Medicine Processing Technology Innovation Center of Hebei Province, Hebei University of Chinese Medicine, Shijiazhuang 050200, China 3. Gansu Institute for Drug Control, Lanzhou 730070, China
To establish the fingerprints of Danggui () and Oudanggui (), and find their differential components using chemometrics methods, thus providing a foundation for identifying the two herbs.A UPLC method was applied to establish the fingerprints. Chromatographic separation was performed on Waters Cortecs T3 column (100 mm × 2.1 mm, 1.6 μm). The mobile phase consisting of 0.1% formic acid acetonitrile solution-0.1% formic acid aqueous solution were adopted for gradient elution with the flow rate of 0.4 mL/min. The column temperature was 35 ℃, and the detection wavelength was at 210 nm. The fingerprints of.and.were evaluated by similarity evaluation, principal component analysis (PCA) and hierarchical cluster analysis (HCA), to find their differential components. At the same time, the main differential components were identified by UPLC-MS/MS and NMR methods.A total of 23 and 19 common peaks were determined from the fingerprints of.and., respectively. Within.group, the similarity of three batches of samples was low, in the range of 0.59—0.80 compared to the common pattern, while the rest was greater than 0.99. Within.group, the similarity of samples varied greatly, in the range of 0.53—0.94. PCA and HCA results showed that the samples of.and.could be clearly divided into two groups. Six chromatographic peaks contributed significantly to the distinction of the two herbs, which were identified as tryptophan, ferulic acid,-ligustilide, isomer of levistilide A, chlorogenic acid and falcarindiol, with the latter two being higher in.and the remaining being higher in..The established UPLC fingerprints combined with the chemometrics method was simple and feasible, which could effectively distinguish.and.as well as reveal their differential components, providing a basis for quality control of.and for establishing the adulteration identification method of.impersonating.. Meanwhile, falcarindiol was isolated from.for the first time, which is the main component of.that distinguishes it from..
(Oliv.) Diels.;Koch.; UPLC fingerprints; chemometrics; falcarindiol; tryptophan; ferulic acid;-ligustilide; isomer of levistilide A; chlorogenic acid
R286.2
A
0253 - 2670(2022)22 - 7214 - 07
10.7501/j.issn.0253-2670.2022.22.026
2022-04-24
王亞丹,女,副研究員,主要從事中藥物質基礎研究及質量控制工作。E-mail: y.dwang@163.com
戴 忠,研究員,主要從事中藥物質基礎研究及質量控制工作。E-mail: daizhong@nifdc.org.cn
姚令文,主任藥師,主要從事中藥物質基礎研究及質量控制工作。E-mail: yaolingwen@nifdc.org.cn
[責任編輯 時圣明]
猜你喜歡
英語世界(2023年10期)2023-11-17 09:19:16
汽車實用技術(2022年10期)2022-06-09 11:16:58
音樂探索(2022年2期)2022-05-30 21:01:37
收藏界(2019年3期)2019-10-10 03:16:40
小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00
中國特種設備安全(2018年11期)2019-01-08 02:08:32
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
中國非營利評論(2017年1期)2017-11-09 03:09:10
海外華文教育(2017年8期)2017-11-07 04:42:02
現代語文(2016年21期)2016-05-25 13:13:50
