基于WGCNA鑒定子宮頸鱗狀細胞癌CD8+T細胞浸潤相關的生物標志物
2022-09-29 07:33:28凡志祥李坤張彥彥左世凱黃冬梅
中國現代醫藥雜志 2022年7期
凡志祥 李坤 張彥彥 左世凱 黃冬梅
子宮頸癌是最常見的婦科惡性腫瘤,其發病率和死亡率在女性惡性腫瘤中位居第四。子宮頸鱗狀細胞癌(Cervical squamous cell carcinoma,CSCC)占子宮頸癌的80%~85%,是最常見的子宮頸癌亞型[1]。近年來,腫瘤免疫檢查點治療成為子宮頸癌治療的研究熱點。然而,尚未發現用于子宮頸癌免疫治療的特異性生物標記物[2]。因此,探索免疫相關生物標記物成為研究子宮頸癌免疫治療的重要方向。檢查點免疫療法已徹底改變了黑色素瘤的治療方式,并在肺癌、腎癌和頭頸癌中顯示出顯著的療效[3~6]。目前,子宮頸癌檢查點免疫治療的試驗尚處于早期階段,而將其作為一種治療選擇主要是因為子宮頸癌是一種病毒驅動的癌癥,免疫系統應將其視為異物[7]。現已證實CD8+T 細胞有助于腫瘤適應性免疫,且在大多數實體瘤中,高度浸潤的CD8+T 細胞對腫瘤治療有益[8,9]。且與正常子宮頸組織相比,子宮頸癌中的CD8+T 細胞更為豐富[10,11]。因此,鑒定與CD8+T 細胞浸潤相關的新生物標志物將有助于探索免疫浸潤機制和豐富免疫治療。
加權基因共表達網絡分析(Weighted gene coexpression network analysis,WGCNA)是一種分析多樣本基因表達譜的方法。它可以通過對具有相似表達模式的基因進行聚類,形成模塊,并分析模塊與特定特征(如患者的臨床信息)之間的關系,該算法已廣泛用于尋找生物標記物[12]。通過估計轉錄本的相對子集進行細胞類型識別(CIBERSORT)是另一種用于分析基因表達數據的生物信息學工具,該工具使用反卷積算法量化免疫細胞的組成,已成功用于估計各種癌癥中的免疫細胞浸潤水平,如乳腺癌和肝癌[13,14]。
為探索腫瘤微環境的特征并確定CSCC 的潛在生物標志物,本研究使用CSCC 的基因表達數據進行WGCNA,并利用CIBERSORT 算法計算樣品的T 細胞組成,然后鑒定出與CD8+T 細胞浸潤水平相關的關鍵模塊和中樞基因。
1 材料與方法
1.1 材料 通過TCGA 數據庫(https://portal.gdc.cancer.gov)獲取304 個原發CSCC 樣本的基因表達譜,并選擇中位絕對偏差前10%的5 605 個基因進行額外分析。從UCSC 數據庫(https://xenabrowser.net/)中檢索285 例CSCC 患者的臨床數據,用于后續分析。
1.2 方法
1.2.1 腫瘤浸潤性免疫細胞(Tumor infiltrating immune cells,TIIC)的評估 使用R 包“CIBERSORT”估算TCGA-CSCC 數據集中每個樣本的22 種TIIC分數[15]。并選擇每個樣本中T 細胞的7 個亞型分數作為WGCNA 的性狀數據。
1.2.2 加權基因共表達網絡的構建 為了解5 605個基因之間的相互關系,并確定基因模塊和影響腫瘤免疫細胞浸潤的中樞基因,使用R Studio 中的“WGCNA”軟件包構建加權基因共表達網絡[16]。首先,基于成對基因之間的Pearson 相關系數,將單個基因的表達水平轉換為相似矩陣。使用表達式矩陣構造一個分層聚類樹來檢測異常值。然后根據網絡拓撲分析函數計算出無標度拓撲擬合指數作為軟閾值功率的函數。接著使用拓撲重疊不相似性度量(TOM)計算具有相似表達譜基因之間的模內連接性。最后采用一種動態混合切割方法將這些基因分成不同的模塊,每個模塊至少有30 個基因。
1.2.3 構建模塊-特征關系 應用Pearson 檢驗法計算模塊特征基因與T 細胞浸潤水平之間的相關性,選擇P值顯著并且相關系數高的T 細胞亞型和模塊,定義其為中心模塊。為確定中心模塊中基因的功能,使用R 包“clusterProfiler”進行富集分析[17]。
1.2.4 hub 基因的識別 根據中心模塊中每個基因的模塊連通性和臨床特征關系選擇候選hub 基因。模塊連通性定義為基因間Pearson 相關性(Module Membership)的絕對值。臨床性狀關系定義為每個基因與性狀之間Pearson 相關性(Gene Significance)的絕對值。將候選hub 基因的Module Membership設置為>0.8,Gene Significance 設置為>0.4。同時選擇中心模塊中的所有基因,應用檢索交互基因的搜索工具(Search Tool for the Retrieval of Interacting Genes,STRING;https://STRING db.org/)構建蛋白質互作(Protein protein interaction,PPI)網絡并尋找中心節點[18]。節點連通性>22 的基因被認為是中心節點。使 用Cytoscape(https:/Cyto-scape.org/)展示網絡并使用R 包“VennDiagram”進行Venn 分析以比較PPI 網絡中的中心節點和候選hub 基因[19]。
1.2.5 hub 基因的驗證 使用TCGA 數據庫驗證hub基因的免疫相關性。首先,根據CIBERSORT 的計算結果提取出每個樣本的CD8+T 細胞比例。使用R 包“ggpubr”計算CD8+T 細胞浸潤水平與hub基因表達之間的Spearman 相關性,并使用R 包“ggstatsplot”對結果進行比較。同時檢索腫瘤-免疫系統相互作用數據庫(Tumor Immune System Interactions Database,TISIDB;http://cis.hku.hk/TISIDB)以確定hub 基因和TIIC 之間的Spearman相關性[20]。最后應用R 包“heatmap”將這些結果以熱圖形式呈現出來。
1.2.6 免疫因子鑒定 hub 基因和不同免疫因子之間的Spearman 相關性來自TISIDB 數據庫,其中包括免疫抑制因子、免疫刺激因子、趨化因子和受體。然后使用R 包“heatmap”構建熱圖,并使用STRING和Cytoscape 選擇與相關免疫因子的平均相關系數大于0.6 的hub 基因構建網絡[19]。
1.2.7 hub 基因的預后分析 通過R 包“survminer”找到hub 基因表達量的最優截斷值,并將患者分為高表達組和低表達組。然后通過R 包“survival”進行Kaplan-Meier 分析。同時采用log-rank 檢驗對hub 基因進行單因素生存分析,當P<0.05 時差異具有統計學意義。并將有統計學意義的hub 基因進行多因素Cox 生存分析,鑒別出對CSCC 預后有獨立影響因素的基因。
1.2.8 基因集富集分析(GSEA) GSEA 是一種基于基因集的富集分析方法[21]。根據基因表達的中值,將樣本分為兩組,并進行“c2.cp.kegg.v7.0.symbols”基因集富集分析,以P<0.05 和q<0.05 作為統計顯著性指標。使用R 包“ggplot2”和“clusterProfiler”對富集途徑進行可視化。在圓圖中,本研究只顯示富集過程的部分核心基因。
2 結果
2.1 CSCC 的加權基因共表達網絡 使用R 包“WGCNA”構建5 605 個基因的共表達網絡,并對304 個CSCC 樣本進行聚類。為構建無標度網絡,選擇β=4(無標度R2=0.85)作為軟閾值功率(見圖1A、1B),并使用動態混合切割技術構建了層次聚類樹,其中樹上的每片葉子代表一個基因,具有相似表達數據的基因靠一起,形成樹的一個分支,代表一個基因模塊,共生成10 個模塊(見圖1C)。
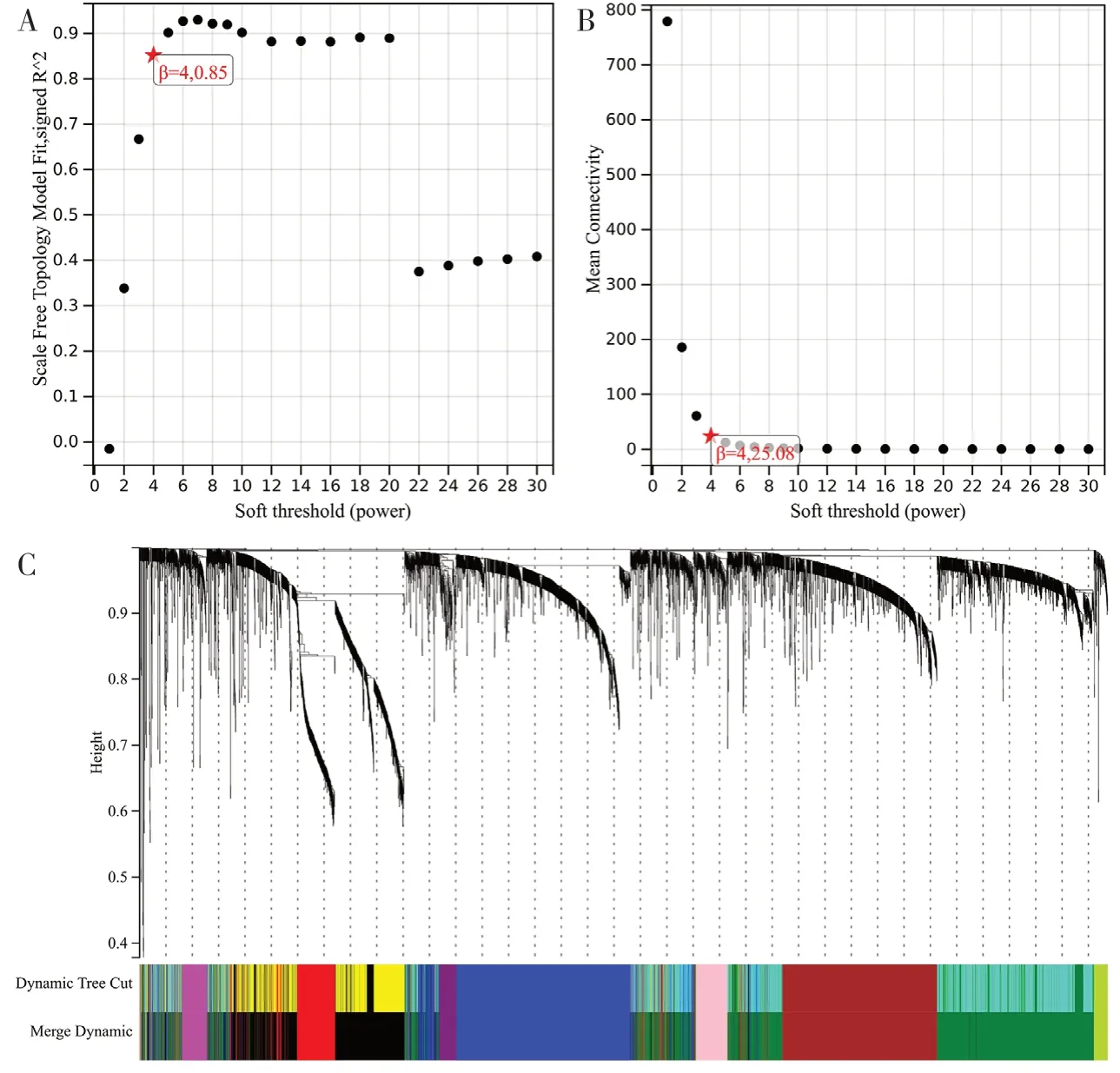
圖1 選擇合適的β值構造層次聚類樹
2.2 識別中心模塊并進行富集分析 模塊特征基因與T 細胞浸潤的相關性見圖2A。在這10 個模塊中黑色模塊與CD8+T 高度相關(r=0.4,P=2.3e-13)。黃綠色模塊與未活化記憶性CD4+T 淋巴細胞也有相對較高的相關性(r=0.33,P=4.6e-9)。其他模塊與T 細胞之間的相關性小于0.3。將與CD8+T 細胞高度相關的黑色模塊作為中心模塊,并使用R 包“GOplot”分析該模塊中所含基因,以進行通路和過程富集分析。前20 個最高富集項中絕大多數與免疫相關(見圖2B),其中前3 個最高富集項為免疫系統的過程(Immune system process)、對刺激的調節反應(Regulation of response to stimulus)和免疫應答(Immune response)。
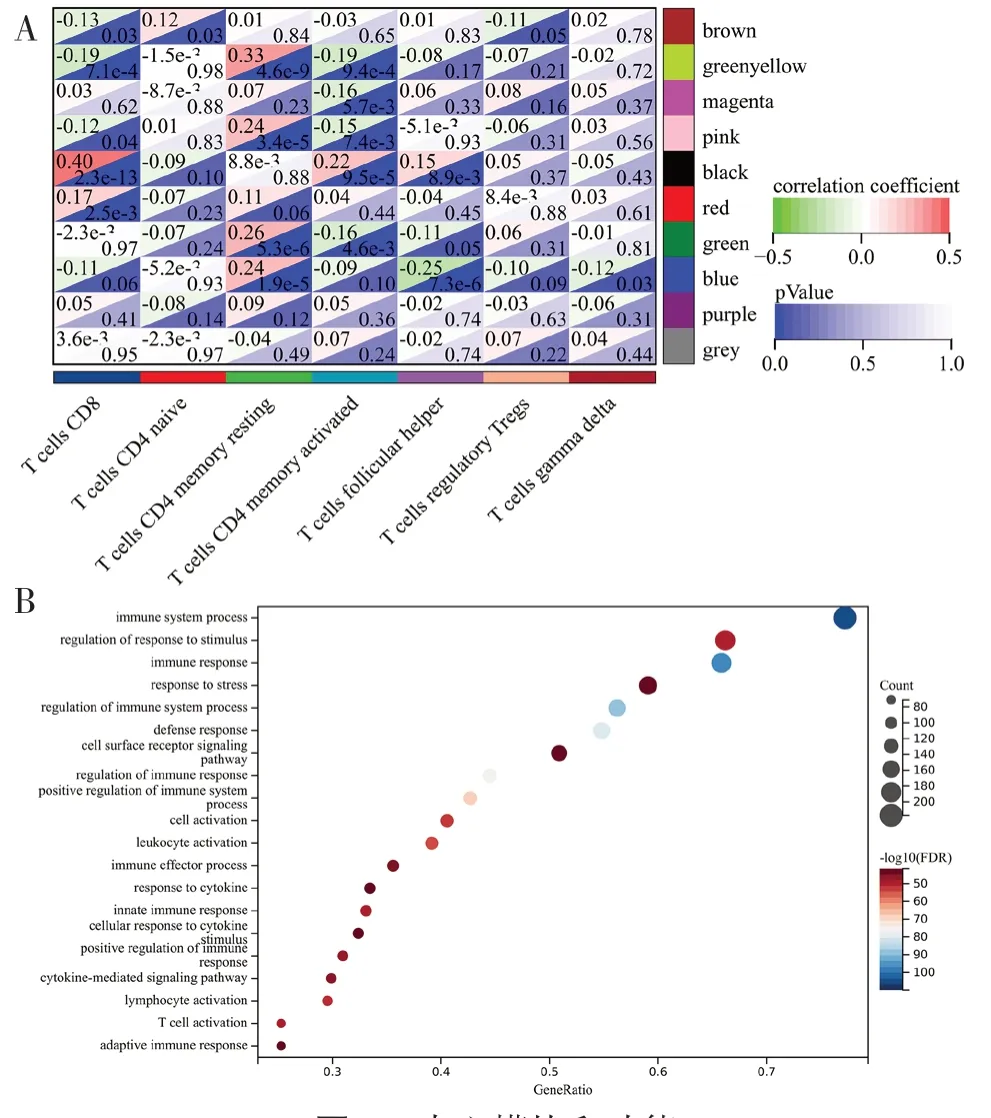
圖2 中心模塊和功能
2.3 hub 基因的識別和驗證 黑色模塊中的高度連接基因被視為與CD8+T 細胞浸潤水平相關的潛在因素。基于PPI 網絡,應用intera-ction score>0.7和degree>22 的截止值(節點)從黑色模塊中確定34 個基因作為中心節點,同時使用Cytoscape將其可視化(見圖3A)。根據閾值標準(Module Membership>0.8 和Gene Significance>0.4),從黑色模塊中篩選68 個基因作為候選hub 基因(見圖3B)。最終選擇在以上兩種方法中均出現的10 個基因為hub 基因(CCL5、CCR5、CD2、CD3D、CD3E、CD8A、CXCR3、IFNG、PDCD1、PRF1),見圖3C。為研究這些hub 基因與CD8+T 細胞之間的關系,使用R 軟件分析TCGA-CSCC 數據集中hub 基因的表達數據與CD8+T 細胞的相關性。結果顯示,10 個基因的mRNA 表達量與CD8+T 細胞的浸潤水平呈顯著正相關(所有基因的相關系數最小為0.47),見圖4A。作為一個示例,圖4B 顯示了CD3E 表達量與CD8+T細胞浸潤水平的關系。查詢TISIDB 數據庫,以獲得腫瘤浸潤淋巴細胞的豐度與hub 基因表達之間的Spearman 相關值。結果顯示hub 基因與腫瘤浸潤淋巴細胞呈正相關。活化CD8+T 細胞(Act CD8)和效應器記憶CD8+T 細胞(Tem CD8)的相關值最高(見圖4C)。此外,以TISIDB 數據庫為基礎,篩選出33 個免疫因子,這些免疫因子與hub 基因的平均相關性大于0.6。基于hub 基因與33 個免疫因子,又構建了免疫浸潤互作網絡,以此來探索hub基因的免疫浸潤機制(見圖4D)。而相關性熱圖詳細展示了hub 基因與不同免疫因子之間的關聯性(見圖5)。

圖3 hub 基因的鑒定
2.4 預后生物標志物的鑒定 使用單因素Cox 回歸分析探索10 個hub 基因與CESC 患者預后之間的關系,發現除PRF1 與IFNG 外,其余8 個hub 基因均與患者預后有顯著的相關性(見圖6A)。而多因素Cox 回歸分析結果表明,僅CCR5(HR=1.61,P=0.03)與CD3E(HR=0.5,P=0.04)與預后顯著相關(見圖6B)。Kaplan-Meier 生存分析結果進一步表明,與低表達組相比,10 個hub 基因高表達組患者有更好的總生存期(OS),見圖7,這與單因素Cox回歸分析結果基本相符。通過觀察發現多因素分析結果顯示CCR5 為CSCC 的危險因素,這與單因素分析和Kaplan-Meier 生存分析的結果完全相反。因此剔除CCR5 后選擇CD3E 作為進一步分析的預后生物標志物。

圖4 hub 基因驗證及蛋白質互作網絡圖構建

圖5 免疫因子和hub 基因之間相關性熱圖

圖6 hub 基因的Cox 回歸分析森林圖

圖7 10 個hub 基的Kaplan-Meier 曲線
2.5 CD3E 的基因集富集分析 根據CD3E 表達中值,將TCGA 中的CSCC 樣本分為高表達組和低表達組進行基因集富集分析。結果表明,CD3E高表達組樣本富集到的免疫相關通路共21 個(FDR<0.05,q<0.05)。其中4 個富集最顯著的途徑分別為細胞黏附分子、趨化因子信號通路、細胞因子-細胞因子受體相互作用和T 細胞受體信號通路(見圖8A、8B)。而低表達組沒有明顯的富集途徑。
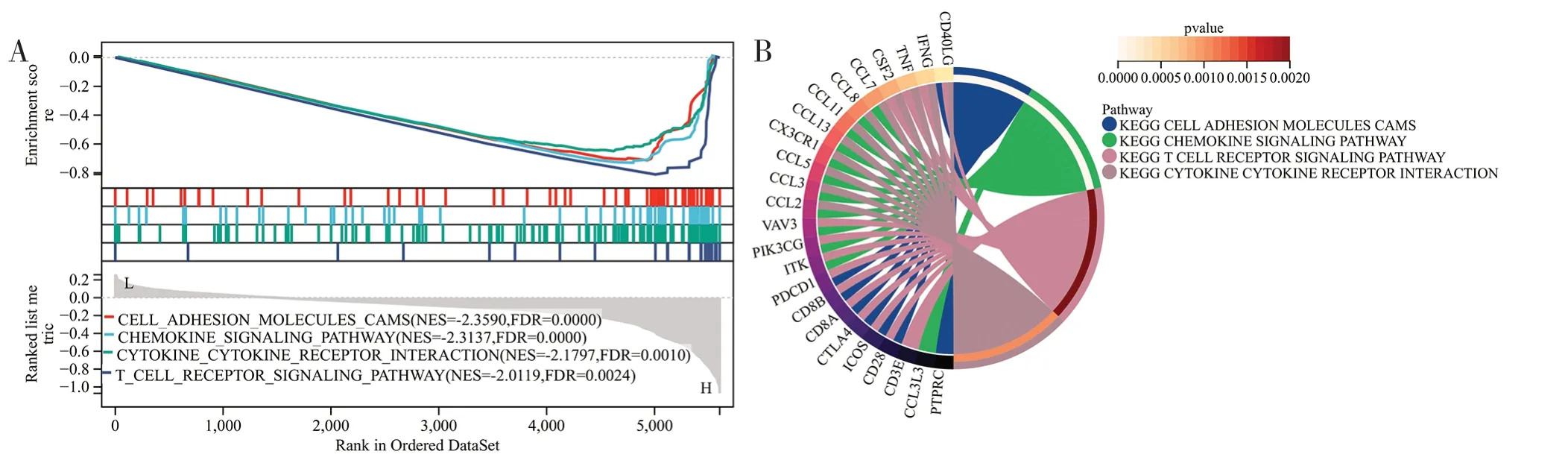
圖8 GSEA 富集分析
3 討論
CSCC 是子宮頸癌的主要組織學亞型,早期CSCC 的預后較好,而對于已發生遠處轉移的晚期子宮頸癌或復發病例,傳統治療手段的效果并不理想[22]。免疫檢查點抑制劑在CSCC 中的成功應用增加了研究人員對探索免疫治療中潛在特異性免疫相關因子的興趣[2]。CD8+T 細胞在免疫治療中起著核心作用。在本研究中,我們鑒定了10 個hub基因,其表達量與CD8+T 細胞浸潤水平相關,提示這些基因可能是影響CESC 免疫浸潤的潛在因素。在這些hub 基因中,CD3E 被確定為CSCC 潛在的預后生物標志物和治療靶點。
目前利用動物腫瘤模型、體外細胞系和臨床樣本技術研究子宮頸癌的分子基礎已取得重大進展。然而,CSCC 具有十分復雜的微環境,需要利用更大的數據集來進一步分析。我們使用基因表達矩陣構建共表達網絡,計算T 細胞的浸潤水平,并確定相關性,以篩選與CD8+T 細胞最相關的基因。對所選模塊的基因集富集分析表明,它是一個與免疫高度相關的模塊。WGCNA 與PPI 網絡之間關聯最緊密的基因被認為是hub 基因(CCL5、CCR5、CD2、CD3D、CD3E、CD8A、CXCR3、IFNG、PDCD1、PRF1)。在TISIDB 數據庫中查詢這10 個基因與免疫細胞之間的關系,發現這些基因的表達與免疫細胞,尤其是CD8+T 細胞顯著正相關。以TISIDB 和STRING 數據庫為基礎,又構建hub 基因和相關免疫因子之間的CD8+T 細胞浸潤網絡,以探索CSCC的免疫機制。分析結果證實hub 基因與CD8+T 細胞浸潤水平密切相關,并在免疫微環境中發揮重要作用。我們還對10 個hub 基因進行了單因素、多因素Cox 回歸分析和Kaplan-Meier 分析。其中CCL5、CCR5、CD2、CD3D、CD3E、CD8A、CXCR3、PDCD1 與CSCC 患者預后有顯著的相關性,且hub基因高表達時患者的預后較好。而多因素Cox 回歸分析僅證實CD3E 低表達是CSCC 不良預后的獨立影響因素。結合這三種分析結果,我們選定CD3E為CSCC 患者潛在的預后指標和治療靶點。
CD3 分子可以與T 細胞受體(TCR)結合,形成CD3/TCR 復合物并介導TCR 信號傳導和T 細胞分化[23,24]。由CD3E 基因編碼的CD3ε是CD3 的一個亞單位,與嚴重免疫缺陷有關,經常被用作CD3 抗體的蛋白質靶點[25,26]。研究發現,CD3E mRNA 表達水平較低的頭頸部鱗狀細胞癌患者復發風險較高[27]。在肝癌、乳腺癌中也觀察到類似的結果,即CD3E 的高表達傾向于有更好的預后[28,29]。GSEA 結果還表明,CD3E 的高表達顯著富集于基質相關信號通路,如T 細胞受體信號通路和趨化因子信號通路。這些結果表明,CD3E 可能還參與了腫瘤微環境從免疫型向代謝型的轉變。因此CD3E 有望成為CSCC 的預后指標和新的免疫治療靶點。
總之,本研究嘗試使用WGCNA 和CIBERSORT算法來識別CSCC 潛在的CD8+T 細胞相關生物標志物,并發現10 個與CSCC 預后相關的hub 基因。通過生物信息學驗證,CD3E 被確定為CSCC 免疫治療的潛在生物標記物和免疫治療靶點。
猜你喜歡
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
財經(2017年15期)2017-07-03 22:40:49
財經(2017年2期)2017-03-10 14:35:35
山東工業技術(2016年15期)2016-12-01 05:31:22
財經(2016年15期)2016-06-03 07:38:02
財經(2016年3期)2016-03-07 07:44:46
財經(2016年6期)2016-02-24 07:41:51
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
終身教育研究(2014年5期)2014-02-28 01:23:06
