新型環狀RNA調控膠質瘤化療耐藥的最新進展Δ
2022-08-11 11:01:44曾昭穆劉超劉麗娜溫稀超何秋果郭巖松鄭克彬河北大學附屬醫院神經外科河北保定07000河北大學臨床醫學院河北保定07000
中國藥房 2022年15期
關鍵詞:耐藥
曾昭穆,劉超,劉麗娜,溫稀超,何秋果,郭巖松,鄭克彬#(.河北大學附屬醫院神經外科,河北 保定 07000;.河北大學臨床醫學院,河北 保定 07000)
膠質瘤是中樞神經系統中最常見的腫瘤之一,占惡性腦腫瘤的75%以上[1]。其中,多形性膠質母細胞瘤被認為是最具侵襲性的原發性惡性腦腫瘤,患者診斷后中位生存期僅為12~15個月[2]。替莫唑胺(temozolomide,TMZ)作為目前最常用的烷化劑類化療藥物,是膠質瘤術后化療的首選方案之一。遺憾的是,以TMZ為基礎的化療方案只能暫時緩解病情的發展,最終還會導致獲得性耐藥和復發,造成患者預后不佳及總體生存期顯著縮短[3]。近年來,大量的研究證實,在膠質瘤耐藥形成過程中存在基因的異常表達,包括編碼和非編碼RNA的差異性表達、致癌基因的激活和抑癌基因的失活等[4]。其中,一種新型非編碼RNA——環狀RNA(circular RNA,circRNA)受到廣泛關注。與傳統線性RNA不同,circRNA的3′和5′末端之間可以形成共價閉合的環狀結構來抵抗核糖核酸酶的剪切,顯現出更強的穩定性、保守性以及組織和細胞特異性[5]。正是基于這些特點,circRNA作為膠質瘤惡性進展過程中的分子靶點,在調控腫瘤增殖、侵襲、遷移和血管生成等方面表現出巨大潛力,已成為膠質瘤診治的潛在標志物[6]。在本文中,筆者從耐藥的角度出發,重點闡述了circRNA調控膠質瘤化療耐藥的分子機制,旨在為開發新的治療方法提供理論依據。
1 膠質瘤化療耐藥機制
既往的研究認為,血腦屏障的存在使得抗腫瘤藥物不能到達腫瘤組織是導致膠質瘤患者化療失敗的根本原因。但2022年發表的一項研究顯示,惡性膠質瘤患者血腦屏障常常存在不同程度的破壞,膠質瘤本身對抗腫瘤藥物的耐受才是導致化療失敗的最本質原因[7]。因此,揭示其分子調節機制對于逆轉腫瘤化療耐藥至關重要。在此,筆者對膠質瘤耐藥機制進行歸納總結。
1.1 藥物轉運代謝
許多抗腫瘤藥物在到達治療靶點后,可被轉運蛋白以逆濃度梯度的形式主動泵出瘤體細胞,進而減少細胞內藥物的積累,導致耐藥發生。這一過程涉及的蛋白主要包括P-糖蛋白(P-glycoprotein,P-gp)、多藥耐藥蛋白(multidrug resistance protein,MRP)、乳腺癌耐藥蛋白(breast cancer resistance protein,BCRP)等[8]。研究發現,P-gp是一種ATP依賴性藥物輸出泵,可同時結合化療藥物和ATP,通過耗能將化療藥物轉移至膠質瘤細胞外,進而使膠質瘤細胞產生耐藥[9]。同時,P-gp也可表達于血管內皮細胞,參與血腫瘤屏障(blood tumour barrier,BTB)引起的耐藥[10]。MRP和P-gp有著相同的作用,可特異性識別疏水性化療藥物的轉運,并與谷胱甘肽結合形成谷胱甘肽巰基共軛物轉運泵,間接轉運弱堿類化療藥物[9-10]。Marinho等[11]在研究中發現,膠質瘤細胞系中均存在MRP1和MRP3的高表達,并且相應基因可以直接調控腫瘤對依托泊苷和長春新堿的耐藥性。BCRP最初分離于耐藥乳腺癌細胞,但其在膠質瘤細胞中也存在高表達,能影響柔紅霉素、米托蒽醌等20余種抗腫瘤藥物的藥效,進而導致膠質瘤細胞產生耐藥性[12]。總體說來,新興研究揭示了膠質瘤放化療耐藥的分子機制,隨著高通量測序技術的快速發展,找到調控藥物轉運代謝的關鍵信號通路,是逆轉膠質瘤細胞化療耐藥的新策略。
1.2 細胞凋亡
當腫瘤細胞出現耐藥時,細胞的凋亡/抗凋亡機制處于失衡狀態。p53是一個經典的抑癌基因,野生型p53可直接誘導腫瘤細胞發生凋亡。在TMZ化療過程中,膠質瘤細胞產生的耐藥機制常表現為野生型p53缺失或突變,O6-甲基鳥嘌呤-DNA甲基轉移酶(O6-methylguanine-DNA methyltransferase,MGMT)的表達明顯增加,進而無法誘導細胞凋亡[13]。B細胞淋巴瘤2(B-cell lymphoma-2,Bcl-2)基因是眾所周知的抗凋亡基因,可抑制腫瘤細胞凋亡而使其產生耐藥,因而也被認定為一種新型的耐藥基因。除此之外,凋亡系統中還有關鍵性因子同源盒(homeobox,HOX)基因,該基因在化療過程中易發生高表達,可通過磷脂酰肌醇3-激酶/蛋白激酶B(phosphoinositide 3-kinase/protein kinase B,PI3K/Akt)信號通路直接抑制腫瘤細胞的凋亡,進而促進腫瘤細胞對TMZ產生耐藥;同時,其還可激活核因子κB(nuclear factor kappa B,NF-κB)信號通路,以此提高腫瘤細胞中MGMT的表達水平,進而減弱化療藥物的細胞毒性[14]。因此,選擇性地針對與腫瘤細胞凋亡相關的基因進行敲除或過表達,這將為膠質瘤化療耐藥的靶向治療帶來新希望。
1.3 DNA損傷修復
破壞腫瘤細胞的DNA結構促使細胞發生凋亡,是最常見的抗腫瘤藥物作用機制之一。反之,在膠質瘤化療過程中,倘若腫瘤細胞修復受損DNA的能力增強,便會導致化療藥物的失效。MGMT作為膠質瘤預后和化療敏感性判斷的指標之一,主要功能是修復由烷化劑造成的細胞內DNA損傷,即MGMT可通過阻止DNA交聯的形成,降低烷化劑對細胞的毒性作用[15]。錯配修復(mismatch repair,MMR)系統主要參與DNA復制錯誤的修復,防止基因發生突變,進而抑制腫瘤的發生發展和耐藥;并且MMR系統與MGMT之間還存在復雜的調控網絡,研究已證明兩者在膠質瘤細胞中呈負相關[13,16]。拓撲異構酶Ⅱ(TopⅡ)也是近年來化療耐藥研究的重要靶點,其可通過降低藥物效應來維持膠質瘤細胞的DNA穩定和基因組完整[17]。此外,另一研究還報道,當多聚ADP-核糖聚合酶 1[poly(ADP-ribose)polymerase-1,PARP-1]、堿基切除修復(base excision repair,BER)蛋白和高遷移率族蛋白A2(high mobility group AT-hook 2,HMGA2)的表達降低時,膠質瘤細胞對TMZ的敏感性均會增強[18]。綜上所述,大量研究證實了DNA損傷修復與膠質瘤化療耐藥存在確切的相關性,對DNA損傷修復相關的作用靶點進行干預修飾,將為逆轉膠質瘤化療耐藥提供新的思路。
1.4 自噬
自噬在腫瘤細胞中可以表現出促進和抑制2種調控機制,這主要取決于腫瘤本身的類型和狀態。當化療藥物刺激腫瘤細胞時,自噬作為一種應激反應被激活,從而通過降解蛋白為腫瘤細胞的代謝提供充足的能量,進而促進腫瘤發展并降低化療藥物的功效[19]。目前研究發現,PI3K/Akt/雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)、缺氧誘導因子1α/C-X-C趨化因子受體 4(hypoxia-inducible factor 1-alpha/C-X-C chemokine receptor type 4,HIF-1α/CXCR4)、Ras/Raf/MEK 等多條信號通路參與TMZ誘導的膠質瘤細胞自噬[20]。因此,調控自噬相關的靶基因來抑制腫瘤耐藥性,可能成為治療膠質瘤耐藥的新策略。
1.5 膠質瘤干細胞變異
膠質瘤干細胞(glioma stem cells,GSCs)存在于膠質瘤組織中,具有超強的增殖分化能力,且常規放化療手段對其基本無效,因此其被認為是膠質瘤復發的“罪魁禍首”[21]。值得注意的是,GSCs具備的這種強大抵抗能力和超長生存時間,使得腫瘤細胞的耐藥性會隨著GSCs變異的積聚而不斷增強。相關研究也證明,膠質瘤化療耐藥與GSCs中多重耐藥基因、ATP結合盒轉運蛋白基因等耐藥基因的高度表達均有密切聯系[22]。綜上所述,精準篩選GSCs中差異表達的基因或蛋白,進一步探究它們的功能及作用機制,將在逆轉膠質瘤化療耐藥方面產生積極作用。
1.6 上皮間充質轉化
目前,越來越多的證據表明,膠質瘤細胞可通過上皮間充質轉化(epithelial mesenchymal transformation,EMT)形成具有GSCs樣特征的細胞;并且GSCs也可以激活EMT相關基因,促進EMT的發生,這2種生物學進程共同造成GSCs數量成倍激增,最終促進膠質瘤侵襲、耐藥等生物學行為的進展[23]。一些學者甚至把EMT、GSCs和化療抵抗統稱為膠質瘤的“邪惡軸心”,認為這是腫瘤難以治愈的根本原因[22]。因此,靶向干預或逆轉EMT為抗膠質瘤化療耐藥提供了新的研究思路。
2 circRNA調控膠質瘤細胞對TMZ耐藥
TMZ作為治療惡性膠質瘤的一線治療藥物被廣泛應用,其細胞毒性作用的主要機制是破壞膠質瘤細胞中的DNA結構,阻止DNA錯配修復,從而誘導腫瘤細胞發生凋亡[3]。目前,已有多項研究證實一些致癌性circRNA參與了膠質瘤細胞對TMZ的耐藥。以circ_ASAP1為例,其在復發性膠質母細胞瘤組織和TMZ抗性細胞系中均顯著上調,其海綿功能可以直接阻礙miR-502-5p與神經母細胞瘤RAS(neuroblastoma RAS,NRAS)的3′-UTR靶向結合,進而促進腫瘤細胞自噬來增強膠質瘤對TMZ的耐藥性[24]。circ_0000936也可以通過競爭性結合miR-1294來促進膠質瘤細胞自噬,從而提高腫瘤細胞的TMZ化療耐藥性[25]。同樣,另一致癌性circ_0076248也被證實可通過促進細胞自噬來降低膠質瘤對TMZ的敏感性,其主要耐藥機制是通過海綿吸附miR-181a來誘導腫瘤細胞中腫瘤蛋白p53和去乙酰化酶1(sirtuin 1,SIRT1)的表達[26]。此外,研究證明,來自TMZ抗性膠質瘤細胞的外泌體可以介導circ_0042003將耐藥性傳遞給對TMZ敏感的膠質瘤細胞[27]。因此,筆者認為外泌體circRNA在耐藥方面具有不可否認的作用,其可以通過調控多種信號通路直接參與膠質瘤化學耐藥的發展。Ding等[28]研究發現,外泌體circ_0072083可靶向結合miR-1252-5p來促進烷基化修復同源蛋白5(alkylation repair homolog protein 5,ALKBH5)介導的去甲基化,進而上調同源框蛋白表達,以此降低TMZ對體外膠質瘤細胞和體內異種移植瘤組織的毒性作用。circ_NFIX也可誘導膠質母細胞瘤細胞對TMZ產生耐藥。Ding等[29]研究證實,外泌體介導的circ_NFIX可以海綿吸附miR-132,進而使受體細胞中MGMT的表達水平顯著升高,最終導致對TMZ敏感的膠質瘤細胞產生耐藥性。
除此之外,還有一些circRNA也被證實可以通過調控凋亡相關基因來誘導膠質瘤細胞對TMZ產生耐藥。由Mcl-1外顯子反向剪接形成的circ_0110757被證實在TMZ耐藥膠質瘤細胞系中表達上調,其潛在耐藥機制是通過miR-1298-5p/整合素α(integrin alpha,ITGα)信號途徑來抑制TMZ誘導的細胞凋亡[30]。同樣,circ_HIPK3通過與miR-524-5p相互作用來刺激膠質瘤細胞中驅動蛋白家族成員2A(kinesin family member 2A,KIF2A)表達上調,從而抑制膠質瘤細胞對TMZ的敏感性和細胞凋亡[31]。circ_0005198是一種在膠質瘤組織、血漿樣本和TMZ耐藥細胞中高表達的競爭性內源RNA,可以靶向結合miR-198來調節三結構域蛋白14(tripartite motif-containing 14,TRIM14)的表達;而降低circ_0005198的表達則可以直接限制耐藥膠質瘤的生物學行為,顯著促進腫瘤細胞凋亡,進而提高細胞對TMZ的敏感性[32]。此外,circRNA還可以靶向ATP結合盒轉運蛋白,通過促進藥物排泄來使腫瘤細胞獲得耐藥性。Hua等[33]的研究證實,miR-145-5p是circ_CEP128的下游靶標,沉默circ_CEP128可以通過促進miR-145-5p與三磷酸腺苷結合轉運蛋白G超家族成員2(ATP binding cassette subfamily G member 2,ABCG2)的靶向結合,來升高膠質瘤細胞內的TMZ濃度,從而增強TMZ的細胞毒性作用。
綜上,circRNA可通過調控膠質瘤細胞中的相關靶基因及信號通路來影響腫瘤細胞對TMZ的耐藥性(具體見表1),這不僅可以給膠質瘤的TMZ治療提供關鍵靶點,還可以為逆轉腫瘤化療耐藥提供新的策略。
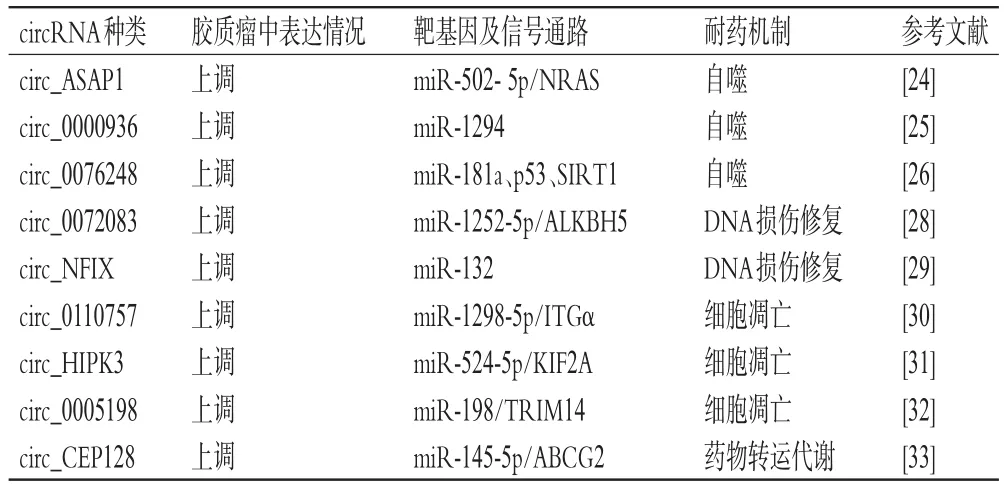
表1 circRNA調控膠質瘤細胞對TMZ耐藥的具體機制
3 circRNA調控膠質瘤細胞對其他抗腫瘤藥物耐藥
在膠質瘤治療過程中,BTB的存在嚴重阻礙了抗腫瘤藥物向中樞神經系統的有效遞送。Gao等[34]報道,circ_USP1在體外膠質瘤腦微血管內皮細胞(gliomaderived microvascular endothelial cell,GDMEC)中呈高表達,并可以作為miRNA的分子海綿與miR-194-5p發生結合。因此,circ_USP1的敲低可以通過介導miR-194-5p/friend白血病病毒整合1(friend leukemia virus integration 1,FLI1)軸直接降低GDMEC中緊密連接相關蛋白5(Cldn-5)、閉合蛋白(Ocln)和閉鎖連接蛋白1(ZO-1)的表達,從而破壞BTB的完整并增加阿霉素的滲透性,最終促進阿霉素誘導膠質瘤細胞凋亡。另一研究發現,RNA結合蛋白KHDRBS3在GDMEC中表達上調,其可與circ_DENND4C結合形成核糖核蛋白復合物,進而提高circ_DENND4C穩定性;同樣,circ_DENND4C作為miR-577的分子海綿,也可影響BTB的通透性。circ_DENND4C的敲低可以顯著增強miR-577對下游靶基因Cldn-5、Ocln和ZO-1的降解,促進阿霉素跨越BTB,最終導致膠質瘤細胞凋亡[35]。circRNA_001160也被認為是GDMEC生長的重要調節劑,可以增強阿霉素耐藥性,其潛在機制是可通過海綿吸附miR-195-5p來上調紅細胞特異性轉化基因變異體1(erythroblast transformation specific variant 1,ETV1)的表達,過表達的ETV1可以與緊密連接相關蛋白的啟動子結合,從而提高緊密連接相關蛋白的表達,最終阻斷阿霉素向膠質瘤細胞的遞送,抑制阿霉素誘導的細胞凋亡[36]。此外,circRNA_104075在膠質瘤對苦參堿耐藥中的作用也已被證實:其可以通過激活Wnt/β-連環蛋白(β-catenin)和PI3K/Akt信號通路來誘導膠質瘤細胞發生自噬,以此減弱苦參堿的細胞毒性[37]。circRNA調控膠質瘤細胞對阿霉素和苦參堿的耐藥機制見表2,其調節機制可為膠質瘤化療耐藥提供新的策略。

表2 circRNA調控膠質瘤細胞對阿霉素和苦參堿耐藥的具體機制
4 總結與展望
circRNA在膠質瘤化療耐藥中起關鍵調控作用,通過使用特異性siRNA或特定過表達載體來靶向糾正耐藥形成過程中內源性circRNA的失調[38],可能是逆轉膠質瘤耐藥的一種有效策略。miRNA類藥物的開發是近年來特別活躍的研究領域,目前已有數百項涉及miRNA類藥物的臨床試驗正在進行之中。例如,Cobomarsen是一種基于鎖核酸修飾的anti-miR-155,其以患者體內的致癌性miR-155為靶點來調節細胞的增殖和分化,目前已進入淋巴瘤和白血病Ⅱ期臨床試驗[39]。遺憾的是,由于circRNA是新興的研究靶點,相關的機制研究仍有待進一步探索,迄今為止,還未見有關于circRNA藥物進入臨床前試驗的報道。但是,circRNA獨特的結構和功能使其作為膠質瘤耐藥治療的潛在靶點仍值得我們深入探索。總之,circRNA已被證明可以直接調控膠質瘤的耐藥性,在監測和克服膠質瘤耐藥方面有著巨大潛力。當前還有許多與耐藥性密切相關的circRNA仍然未知,仍迫切需要我們進行更深入的科學研究和臨床試驗,以進一步開發其臨床價值。
猜你喜歡
保健醫苑(2022年5期)2022-06-10 07:46:38
現代臨床醫學(2022年3期)2022-06-06 07:59:40
昆明醫科大學學報(2022年1期)2022-02-28 07:43:40
天津醫科大學學報(2021年3期)2021-07-21 09:04:02
科學大眾(2020年12期)2020-08-13 03:22:22
云南醫藥(2019年3期)2019-07-25 07:25:10
現代檢驗醫學雜志(2016年1期)2016-11-12 13:19:40
國外醫藥(抗生素分冊)(2016年6期)2016-07-10 11:34:45
中國衛生標準管理(2015年14期)2016-01-15 02:58:37
中國當代醫藥(2015年17期)2015-03-01 02:03:58
