新型多羧酸衍生物的合成、線路優化與熒光性質*
2022-07-29 00:56:40尉祎雯馮凱茵趙天賜
化工科技 2022年2期
尉祎雯,馮凱茵,趙天賜,鄭 蕾,成 昭
(西安醫學院 藥學院,陜西 西安 710021)
20世紀80年代初,錢永健教授課題組對二(β-氨基乙基)乙二醇醚-N,N,N′,N′-四乙酸(Ethylene Glycol Tetraacetic Acid,EGTA)配體進行結構修飾,以苯環取代EGTA結構中N、O之間橋連的2個亞甲基,合成得到了一系列新型配體1,2-二(2-氨基苯氧基)乙烷-N,N,N′,N′-四乙酸[1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid,BAPTA][1-3](見圖1)及其衍生物,并在BAPTA配體結構上進一步連接熒光基團,相繼得到了一系列對金屬離子表現特征識別性能的小分子BAPTA受體熒光探針。
作為金屬離子配體,EGTA與BAPTA的4條羧酸鏈能夠形成空間環狀結構,當環的空腔尺寸與目標離子匹配時[4-5],即可發生EGTA、BAPTA與目標離子的配位與識別作用。此外,作為一種苯環上連接四羧酸結構的給電子受體,BAPTA型受體對離子的識別作用,受到其苯環電子云密度的影響[6-7]。錢永健教授在進行BAPTA受體的結構設計時,還通過改變BAPTA苯環上的取代基,引入吸電子、給電子取代基,改變苯環電子云密度,進而調節BAPTA受體與離子結合能力的強弱。在Biochemistry發表的代表性工作中[1],錢永健教授課題組首次在BAPTA苯環中分別引入給電子取代基—CH3和吸電子取代基—Br,并測定發現,以—CH3取代BAPTA苯環上的—H,可使BAPTA對金屬離子的結合強度增加4個log單位,而—Br對上述—H的取代,則使BAPTA對金屬離子的結合強度降低1.2個log單位。
作者設計合成BAPTA衍生物2-[2-(5-甲基-2-氨基苯氧基)乙氧基]-4-苯并咪唑基苯胺-N,N,N′,N′-四乙酸甲酯(S12)時,亦在BAPTA苯環引入4′—CH3,期望增加BAPTA受體部分電子云的密度、調節其配位性能。作為最小的烷基與給電子取代基,甲基的引入不會造成較大空間位阻,且幾乎不影響四羧酸環狀結構、不影響受體結合底物的選擇性與作用方式[8]。在BAPTA經典合成方法的基礎上,進行其中幾步反應的合成條件優化,提高了產物收率與反應的選擇性,并進一步研究了目標產物S12的紫外、熒光性質。
1 實驗部分
1.1 試劑與儀器
鄰硝基酚、5-甲基-2-硝基苯酚、1-溴-2-氯乙烷、溴乙酸甲酯、二異丙基乙胺、鄰苯二胺、無水碳酸鉀、還原鐵粉:國藥集團化學試劑有限公司;N,N-二甲基甲酰胺(DMF)、乙腈、甲醇、無水乙醇、乙酸乙酯、石油醚:天津市天力化學試劑有限公司;以上試劑均為化學純。
顯微熔點儀:XT-4,北京泰克科技有限公司;傅里葉變換紅外光譜儀:TENSOR T-27,超導核磁共振波譜儀:AVANCE Ⅲ 400MHz,飛行時間-質聯儀:microTOF-QⅡ ESI-Q-TOF LC/MS/MS,美國Bruker公司;紫外分光光度計:UV-1700,日本SHIMADZU制作所;熒光分光光度計:LS-55,美國PerkinElmer公司。
1.2 實驗方法
以鄰硝基酚、5-甲基-2-硝基苯酚、1-溴-2-氯乙烷、溴乙酸甲酯、鄰苯二胺等為原料,經6步合成得到一種BAPTA型熒光探針S12,合成路線見圖2。

圖2 化合物S12的合成路線
1.2.1 2-(2-氯乙氧基)-硝基苯(化合物1)的合成

1.2.2 4-甲基-2-[2-(2-硝基苯氧基)乙氧基]硝基苯(化合物2)的合成
1.2.3 4-甲基-2-[2-(2-氨基苯氧基)乙氧基]苯胺(化合物3)的合成
1.2.4 4-甲基-2-[2-(2-氨基苯氧基)乙氧基]苯胺-N,N,N′,N′-四乙酸甲酯(化合物4)的合成
1.2.5 2-[2-(5-甲基-2-氨基苯氧基)乙氧基]-4-甲酰基苯胺-N,N,N′,N′-四乙酸甲酯(化合物5)的合成
1.2.6 S12的合成
1.3 結構表征
IR:采用溴化鉀壓片法對各步合成產物進行IR測試,波數為4 000~400 cm-1。
1H NMR:以四甲基硅為內標、CDCl3為溶劑,對各步合成產物分別進行400 MHz的1H NMR測試。
熒光分析:出射與入射狹縫寬度均為1 nm,波長為200~500 nm。
2 結果與討論
2.1 表征結果
合成中間產物的結構經IR與1H NMR測定,表征數據均與預期結構相符。另由探針S12的1H NMR、IR與MS等表征數據可知,探針S12結構正確,經合成引入的官能團羰基、醚鍵等均呈現其特征吸收,特征氫位移均能由1H NMR進行歸屬,符合實驗預期結構設計,可以進行進一步的光學性質研究。
2.2 2-(2-氯乙氧基)-硝基苯(化合物1)的合成路線優化
文獻報道2-(2-氯乙氧基)-硝基苯的合成方法[6],多使用1,2-二溴乙烷、原料鄰硝基酚的鉀鹽或鈉鹽作為反應物(見圖3)[9],但1,2-二溴乙烷中2個溴原子的選擇性不強,除生成目標產物2-(2-氯乙氧基)-硝基苯(見圖3化合物a),反應常伴隨著生成2分子鄰硝基酚偶聯的副產物b,致使產率降低、副產物難于分離。
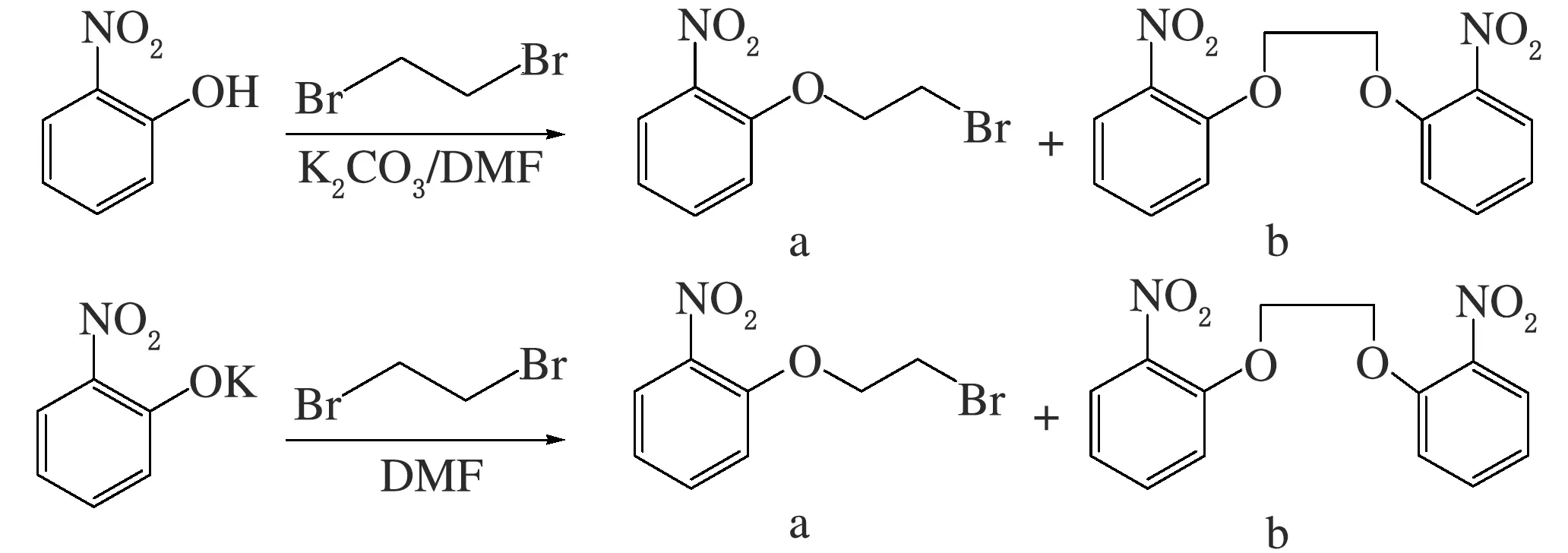
圖3 2-(2-氯乙氧基)-硝基苯的經典合成路線
為避免副產物b的生成,選擇了具有反應活性差異的不對稱試劑1-溴-2-氯乙烷,替代對稱試劑1,2-二溴乙烷,—Br與—Cl的反應活性差異提供了更高的反應位點選擇性,目標產物2-(2-氯乙氧基)-硝基苯(化合物1)的合成與后續—Cl發生取代反應、得到4-甲基-2-[2-(2-硝基苯氧基)乙氧基]硝基苯(化合物2)的反應收率均高于95%,且產物后處理簡單,實現了對2-(2-氯乙氧基)-硝基苯(化合物1)(見圖4)與后續產物4-甲基-2-[2-(2-硝基苯氧基)乙氧基]硝基苯(化合物2)合成路線的極大優化。

圖4 2-(2-氯乙氧基)-硝基苯的優化合成路線
2.3 4-甲基-2-[2-(2-氨基苯氧基)乙氧基]苯胺(化合物3)的合成方法優化
4-甲基-2-[2-(2-氨基苯氧基)乙氧基]苯胺(化合物3)的合成中,以平行實驗篩選合成方法、優化實驗方案,反應物4-甲基-2-[2-(2-硝基苯氧基)乙氧基]硝基苯(化合物2)的加入量均控制為3.18 g、0.01 mol,溶劑均為無水乙醇,各方法及其產物收率見表1。
A法[10-11]先進行反應物與鐵粉的混合、再加入鹽酸,因鐵粉未完全活化,致使產率較低。C法[12]中加入鹽酸進行鐵粉活化、但過量鹽酸產生了過多鐵粉的消耗,產率仍不理想。以醋酸作為反應引發介質的B法[13],因醋酸的酸性較弱、速率較慢,反應效果受限。D法合成路徑中,進行鐵粉充分活化與反應物分批投料,優化合成條件、提高產率,先進行少量鹽酸與鐵粉的混合、微熱(將少量鹽酸加入鐵粉中,加熱下攪拌反應15~30 min),使鐵粉表面充分活化,再于30 min內分批加入反應物,使鐵粉與分批次加入的反應物充分接觸、充分反應,實現了產物收率的大幅提高。

表1 鐵粉還原反應的條件篩選
2.4 探針S12的熒光激發與發射光譜
在200~500 nm進行濃度為1×10-5mol/L探針S12溶液的紫外-可見與熒光光譜掃描[14-15],見圖5。
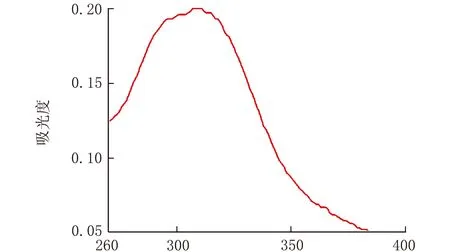
λ/nma 紫外-可見光譜
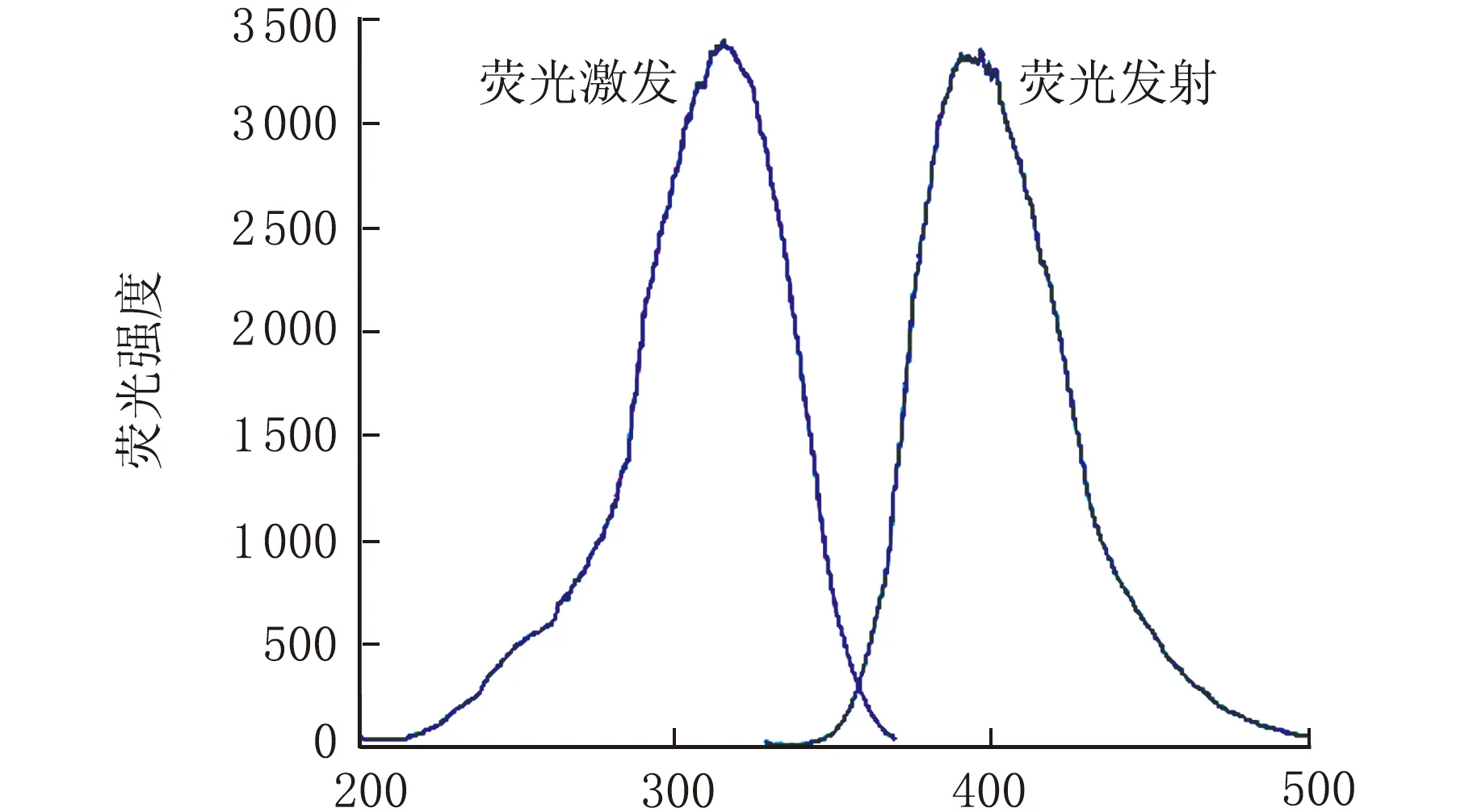
λ/nmb 熒光激發與發射光譜圖5 S12的紫外-可見光譜和熒光激發與發射光譜
由圖5a可知,S12的最大吸收波長位于314 nm。由圖5b可知,S12熒光激發與發射光譜呈現良好的熒光強度與鏡像對稱關系[16-17](λex=313 nm,λem=397 nm),呈現較大的斯托克位移,為84 nm。圖5aS12紫外-可見光譜吸收峰314 nm與圖5b熒光激發光譜的峰值波長λex=313 nm基本保持一致,光學性質穩定。
3 結 論
基于BAPTA配體的經典合成路線,在BAPTA苯環引入4′—CH3,合成得到一種BAPTA衍生物S12。給電子基—CH3的引入,增加了BAPTA受體部分電子云的密度,光譜研究顯示,BAPTA衍生物S12,其紫外-可見與熒光光譜出現最大吸收與發射波長向長波方向的紅移。此外,進行BAPTA經典合成路徑中關鍵反應步驟的條件優化,有效提高了產物收率與反應的選擇性,且產物后處理簡單,提高了多步合成路線整體的原子利用率,為實現合成路徑的綠色化邁進了一步。
