多中心切割二維液相色譜法同時測定九味熄風顆粒中10種成分
2022-07-21 02:36:18沈燦杰陳夏霖王振中
中草藥 2022年14期
沈燦杰,馬 陽,陳夏霖,高 霞,曹 亮,王振中,肖 偉
多中心切割二維液相色譜法同時測定九味熄風顆粒中10種成分
沈燦杰1, 2, 3,馬 陽1, 2,陳夏霖1, 2,高 霞1, 2,曹 亮1, 2,王振中1, 2,肖 偉1, 2*
1. 江蘇康緣藥業股份有限公司,江蘇 連云港 222001 2. 中藥制藥過程新技術國家重點實驗室,江蘇 連云港 222001 3. 中國藥科大學,江蘇 南京 211198
采用在線多中心切割二維液相色譜法,建立同時測定九味熄風顆粒中10種成分的含量測定方法。第一維采用Acquity BEH C18(100 mm×3.0 mm,1.7 μm)為色譜柱,第二維色譜柱為X Select HSS T3(250 mm×4.6 mm,5 μm);流動相均采用甲醇-乙腈(95∶5)混合溶液-磷酸水體系,梯度洗脫;體積流量分別為0.2、1.0 mL/min;柱溫35 ℃。同時測定了九味熄風顆粒中次黃嘌呤、天麻素、有機酸類(原兒茶酸、新綠原酸、綠原酸、隱綠原酸)、環烯醚萜苷類(馬錢苷酸、獐芽菜苦苷、龍膽苦苷、當藥苷)10種成分的含量,各成分在考察的質量濃度范圍內線性關系良好(≥0.999),精密度RSD均小于2%,重復性RSD均小于5%,供試品溶液室溫24 h內穩定,平均加樣回收率為97.45%~104.82%,13批制劑中次黃嘌呤、天麻素、原兒茶酸、新綠原酸、綠原酸、馬錢苷酸、隱綠原酸、獐芽菜苦苷、龍膽苦苷、當藥苷質量分數分別為0.034~0.111、1.224~2.078、0.032~0.055、0.010~0.066、0.202~0.341、1.052~1.542、0.016~0.080、0.151~0.333、4.560~7.354、0.343~0.518 mg/g,其中天麻素、總有機酸、總環烯醚萜苷類成分的批間一致性良好。建立的方法操作簡便,結果準確、可靠,可用于九味熄風顆粒的質量控制和評價。
九味熄風顆粒;二維液相色譜;中心切割;同時測定;次黃嘌呤;天麻素;有機酸類;原兒茶酸;新綠原酸;綠原酸;隱綠原酸;環烯醚萜苷類;馬錢苷酸;獐芽菜苦苷;龍膽苦苷;當藥苷;質量控制
九味熄風顆粒(Jiuwei Xifeng Granules,JXG)是由江蘇康緣藥業股份有限公司研制生產的中藥復方制劑,由天麻、熟地黃、鉤藤、龍膽、法半夏、僵蠶、龜甲、龍骨、青礞石9味藥材組成,具有滋陰平肝、熄風化痰的功效[1],臨床上用于輕中度小兒多發性抽動癥,證屬腎陰虧損、肝風內動證者[2]。多指標的定量測定對該制劑的質量控制、保證臨床療效具有重要意義。然而JXG成分復雜,含有生物堿、環烯醚萜、有機酸等多類活性成分,進行一維色譜分析時,樣品中目標成分多存在共流出峰,或達不到基線分離,無法對目標分析物實現同時定量分析。
近年來,多維液相色譜分離成為液相色譜發展的一個重要方向,它將不同性能或特點的色譜體系進行聯用,具有高分離效果和高峰容量的特點[3],《中國藥典》2020年版中也新增了多維液相分析技術[4]。采用2個合適的色譜聯用(二維液相色譜)可滿足大多數難分離樣品的分離要求。二維液相色譜根據切割全部還是部分的一維洗脫液進入二維分離,可分為全二維色譜(LC×LC)和中心切割色譜(LC-LC)[4-5]。二者中的選擇性全二維(sLC×LC)和多中心切割式二維(mLC-LC)可以解決多個峰重疊的難題,對多個目標成分進行定量分析[6],已經廣泛用于分析食品、環境、中藥、生物樣本等復雜基質樣品或手性化合物等難分離混合物[7-13]。二維多中心切割模式一般通過閥的切換將第一維中感興趣的部分依次切割,臨時儲存在定量環或捕獲柱或者直接轉移進入選擇性不同的第二維進行分離[14-15]。
本實驗基于二維液相多中心切割技術,成功建立了JXG中多成分的快速分析方法。此方法第一維對綠原酸、隱綠原酸、馬錢苷酸、龍膽苦苷、當藥苷進行分析,第二維根據次黃嘌呤、天麻素、原兒茶酸、新綠原酸、獐芽菜苦苷的一維出峰時間,設置切割時間窗口,在第二維色譜柱上進行進一步分離,實現對制劑中10種成分進行含量測定。
1 儀器與材料
Agilent 1290 Infinity II二維液相色譜系統,配備2臺1290 Infinity II高速泵,1290 Infinity II自動進樣器,1290 Infinity II柱溫箱以及2臺1290 Infinity II二極管陣列檢測器,外置2位/4通雙向閥及閥驅動,美國Agilent公司;KH-300DB型數控超聲波清洗器,昆山禾創超聲波儀器有限公司;Pacific TII7超純水儀,美國Thermo公司;ME104E型電子天平、XS205DU型電子天平,Mettler Toledo公司。
對照品天麻素(質量分數96.8%,批號110807-201306)、原兒茶酸(質量分數99.9%,批號110809-201205)、綠原酸(質量分數99.3%,批號110753-201716)、馬錢苷酸(質量分數97.4%,批號11865-201704)、獐芽菜苦苷(質量分數98.3%,批號110785-201404)、龍膽苦苷(質量分數97.1%,批號110770-201918)、當藥苷(質量分數100%,批號111742-200501)均購自中國食品藥品檢定研究院;對照品次黃嘌呤(質量分數99.3%,批號6057)、新綠原酸(質量分數99.4%,批號4974)均購自上海詩丹德生物技術有限公司;對照品隱綠原酸(質量分數98.0%,批號15121702),購自成都普菲德生物技術有限公司。
JXG(6 g/袋),江蘇康緣藥業股份有限公司,批號190102、190103、190701、190702、190901、190902、191002、191003、200501、200503、210301、210302、210303,分別編號為S1~S13。乙腈、甲醇,色譜純,美國Tedia公司;無水甲醇、磷酸,分析純,南京化學試劑有限公司;水為超純水。
2 方法與結果
2.1 對照品溶液制備
取次黃嘌呤、天麻素、原兒茶酸、新綠原酸、綠原酸、馬錢苷酸、隱綠原酸、獐芽菜苦苷、龍膽苦苷、當藥苷對照品適量,精密稱定,加50%甲醇制成質量濃度分別為25.6、701.8、15.9、32.3、97.7、420.3、33.7、61.4、2 502.8、277.8 μg/mL的混合對照品儲備液。精密移取1 mL置10 mL量瓶,加50%甲醇稀釋至刻度,即得混合對照品溶液。
2.2 供試品溶液制備
取JXG適量,研細,取約1 g,精密稱定,置具塞錐形瓶中,精密加入50%甲醇25 mL,密塞,稱定質量,超聲處理(頻率40 kHz、功率500 W)30 min,放冷,再稱定質量,用50%甲醇補足減失的質量,搖勻,濾過,取續濾液,即得供試品溶液。
2.3 陰性對照樣品溶液的制備
按JXG處方比例和工藝,分別制備缺少天麻、半夏藥材的陰性對照樣品;缺少龍膽藥材的陰性對照樣品;缺少僵蠶藥材的陰性對照樣品;缺少鉤藤藥材的陰性對照樣品;并按“2.2”項下方法制備各陰性對照樣品溶液。
2.4 色譜條件及系統適用性試驗
2.4.1 一維色譜條件 以Acquity UPLC BEH C18(100 mm×3 mm,1.7 μm)為色譜柱;柱溫35 ℃;體積流量0.2 mL/min;檢測波長為220、250、327 nm;以0.05%磷酸水溶液為流動相A,甲醇-乙腈(95∶5)混合溶液為流動相B,梯度洗脫程序:0~6 min,1%~1.75% B;6~8 min,1.75%~6% B;8~16 min,6%~12% B;16~35 min,12% B;35~40 min,12%~20% B;40~50 min,20%~27% B;50~51 min,27%~95% B;51~58 min,95% B;58~60 min,95%~1% B;進樣體積1 μL。
2.4.2 二維色譜條件 色譜柱為XSelect HSS T3柱(250 mm×4.6 mm,5 μm);柱溫35 ℃;體積流量1.0 mL/min;檢測波長:220、250、327 nm;流動相A為0.015%磷酸水溶液,流動相B為甲醇-乙腈(95∶5)混合溶液。梯度洗脫程序:0~6 min,0 B;6~13 min,0~1% B;13~15 min,1% B;15~25 min,1%~12% B;25~35 min,12%~21% B;35~42 min,21%~30% B;42~51 min,30%~40% B;51~52 min,40%~95% B;52~59 min,95% B;59~60 min,95%~0 B。
2.4.3 系統流路 系統流路連接見圖1,其中接口采用2位/4通雙向閥可以實現流路的完全對稱,減小閥切換時系統壓力的變化[16]。閥在位置A時,組分經第一維洗脫進入廢液,至目標組分時閥切換到位置B,目標組分暫時儲存于定量環中;然后閥切換至位置A,定量環中的切割組分隨第二維流動相進入第二維色譜柱進行分析,至下一次切割時閥切換回位置B。
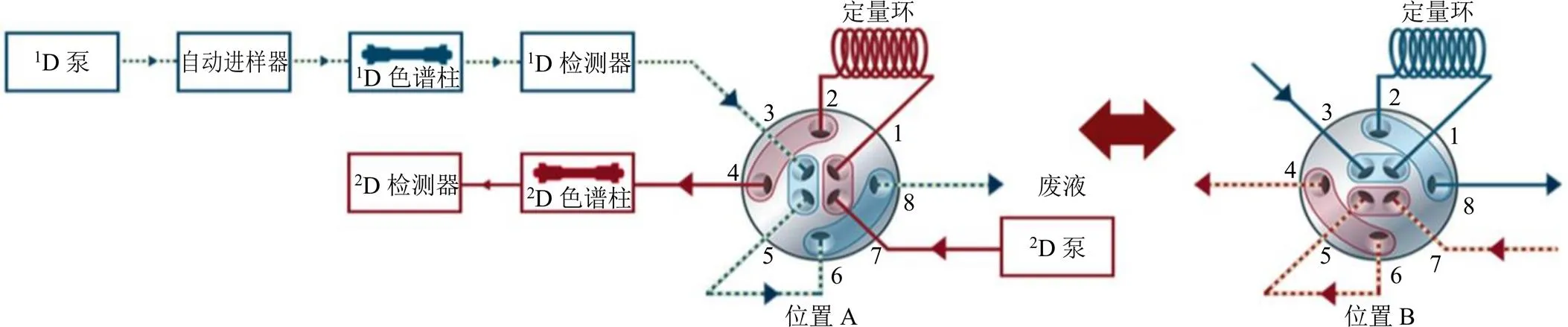
圖1 多中心切割二維液相系統連接示意圖
2.4.4 二維切割時間 考慮到一維液相各色譜峰的峰寬差異,保證不造成切割過程中的損失,選擇200 μL定量環進行儲存,在一維體積流量為0.2 mL/min條件下收集時間為0.8 min。閥切換時間為0 min(位置A);4.10 min(位置B),4.90 min(位置A);12.00 min(位置B),12.80 min(位置A);14.85 min(位置B),15.65 min(位置A);19.00 min(位置B),19.80 min(位置A);42.00 min(位置B),42.80 min(位置A)。
2.4.5 系統適用性試驗 精密移取混合對照品溶液和供試品樣品溶液適量,按照本色譜條件測定,對照品和供試品溶液的一維及二維分離色譜圖如圖2所示。本方法第一維在250 nm波長下對馬錢苷酸、龍膽苦苷、當藥苷進行分析,在327 nm下對隱綠原酸、綠原酸進行分析;第二維在220 nm波長下對天麻素、原兒茶酸進行分析,在250 nm下對次黃嘌呤、獐芽菜苦苷進行分析,在327 nm下對新綠原酸進行分析。在60 min內一維和二維色譜圖基線平穩,各成分分布均勻,與相鄰色譜峰的分離度均不低于1.5,拖尾因子在0.8~1.5,理論塔板數以各色譜峰計均在4000以上,可進行準確定量。
2.5 專屬性試驗
精密移取“2.1~2.3”項下的各溶液適量,按照本色譜條件測定,結果見圖3。各陰性樣品溶液在所測成分相應的位置處未見色譜峰,說明陰性樣品溶液對測定成分沒有干擾。
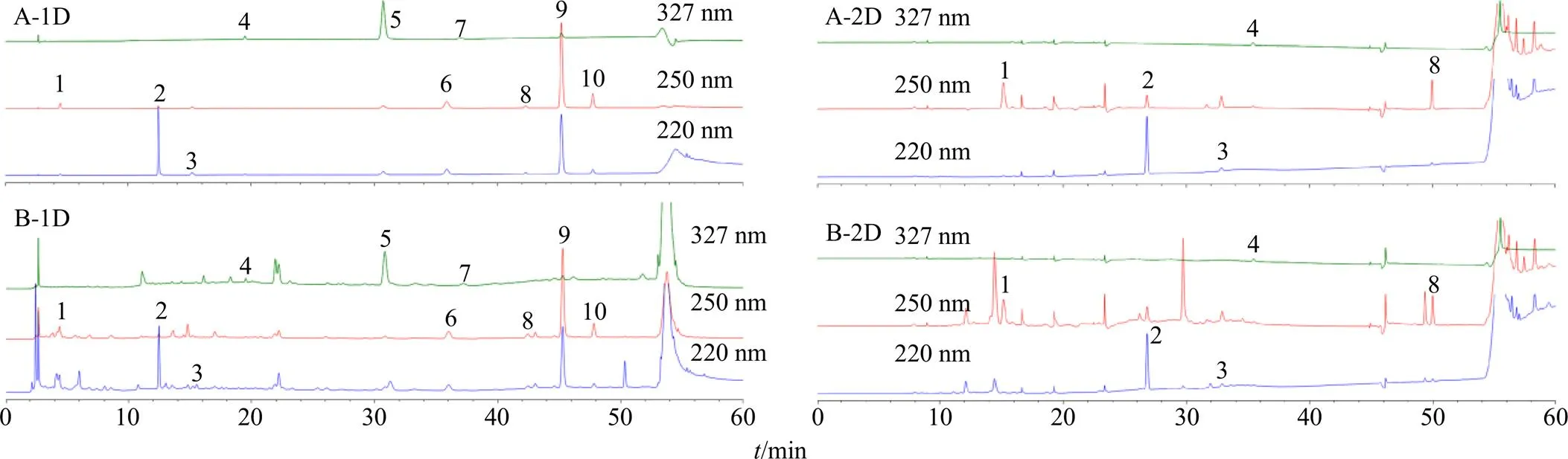
1-次黃嘌呤 2-天麻素 3-原兒茶酸 4-新綠原酸 5-綠原酸 6-馬錢苷酸 7-隱綠原酸 8-獐芽菜苦苷 9-龍膽苦苷 10-當藥苷,下圖同
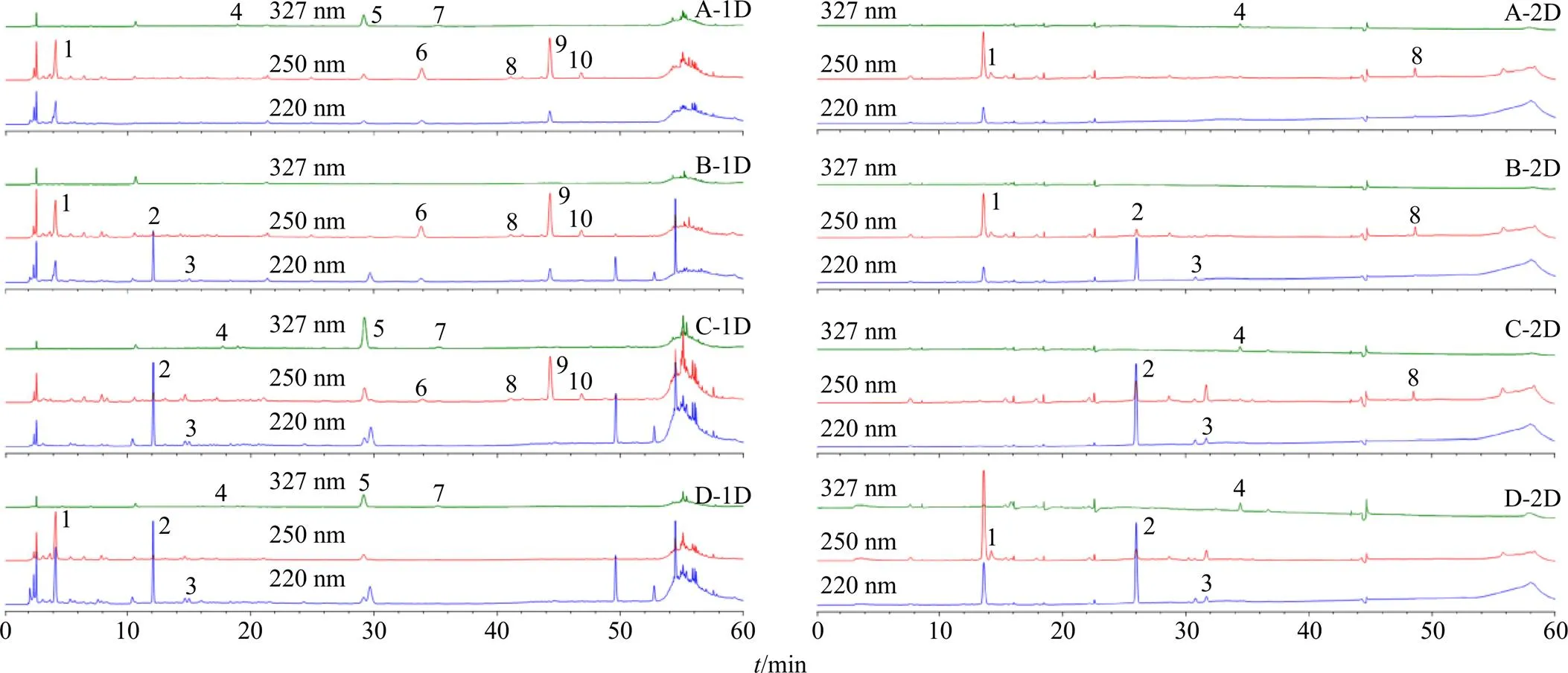
圖3 缺天麻和半夏(A)、缺鉤藤(B)、缺僵蠶(C)、缺龍膽(D) 陰性樣品的一維(1D) 和二維(2D) 分離色譜圖
2.6 線性關系考察
精密移取“2.1”項下混合對照品儲備液,50%甲醇逐級稀釋,制得系列質量濃度對照品溶液。按“2.3”項下色譜條件進樣分析,記錄各色譜峰的峰面積。以對照品質量濃度為橫坐標(),峰面積為縱坐標(),繪制標準曲線,以信噪比不小于10確定目標化合物的定量限,10種成分的回歸方程、線性范圍與定量限見表1。
2.7 精密度試驗
取“2.1”項下混合對照品溶液,按“2.3”項下色譜條件進行測定峰面積,連續進樣6次,結果次黃嘌呤、天麻素、原兒茶酸、新綠原酸、綠原酸、馬錢苷酸、隱綠原酸、獐芽菜苦苷、龍膽苦苷、當藥苷峰面積的RSD分別為0.19%、0.13%、1.03%、1.59%、0.29%、0.29%、1.09%、0.33%、0.15%、0.16%,表明方法精密度較好。
表1 10種成分的回歸方程、線性范圍與定量限
Table 1 Regression equation, linear range, and limits of quantification of 10 components
成分線性方程r線性范圍/(μg?mL?1)定量限/(μg?mL?1) 次黃嘌呤y=25.462 x+0.169 30.999 80.51~5.110.26 天麻素y=10.738 x+3.004 60.999 814.04~140.360.56 原兒茶酸y=33.069 x-0.201 70.999 70.32~3.180.22 新綠原酸y=18.619 x+0.066 70.999 80.32~3.230.19 綠原酸y=13.658 x-0.631 20.999 71.95~19.540.49 馬錢苷酸y=4.899 4 x+0.679 40.999 98.41~84.051.12 隱綠原酸y=13.214 x+0.470 80.999 70.34~3.370.23 獐芽菜苦苷y=6.902 x-0.325 90.999 81.23~12.290.46 龍膽苦苷y=6.293 x+10.230.999 750.06~500.550.63 當藥苷y=8.573 9 x+1.142 60.999 95.56~55.550.37
2.8 穩定性試驗
取JXG(S4)約1 g,精密稱定,按“2.2”項下方法制備供試品溶液,分別室溫放置0、2、4、8、12、18、24 h,按“2.3”項下色譜條件進行測定,記錄各待測成分的峰面積,結果供試品溶液中次黃嘌呤、天麻素、原兒茶酸、新綠原酸、綠原酸、馬錢苷酸、隱綠原酸、獐芽菜苦苷、龍膽苦苷、當藥苷的峰面積RSD分別為0.41%、0.73%、1.38%、1.90%、0.34%、0.50%、1.69%、1.10%、0.32%、0.31%,均小于2%,表明供試品溶液在24 h內穩定。
2.9 重復性試驗
分別取同一批號的JXG(S4)約1 g,精密稱定,按“2.2”項下方法制備供試品溶液,平行制備6份,取“2.1”項下混合對照品溶液,按“2.3”項下色譜條件進行測定,按外標法計算各待測成分的含量及RSD。計算得到次黃嘌呤、天麻素、原兒茶酸、新綠原酸、綠原酸、馬錢苷酸、隱綠原酸、獐芽菜苦苷、龍膽苦苷、當藥苷的平均質量分數分別為0.056、1.574、0.032、0.010、0.202、1.069、0.016、0.151、5.893、0.638 mg/g,RSD分別為1.55%、0.66%、2.53%、2.10%、0.93%、0.66%、1.88%、1.61%、0.94%、0.42%,結果表明該方法重復性較好。
2.10 加樣回收率試驗
取已測知含量的樣品(S4)9份,每份約0.5 g,精密稱定,分成3組,分別加入相當于樣品中待測成分質量分數的50%、100%、150% 3個水平的混合對照品溶液,按“2.2”項下方法制備,即得低(50%)、中(100%)、高(150%)質量濃度的回收率供試品溶液,測定含量,計算各成分的加樣回收率,結果次黃嘌呤、天麻素、原兒茶酸、新綠原酸、綠原酸、馬錢苷酸、隱綠原酸、獐芽菜苦苷、龍膽苦苷、當藥苷的平均加樣回收率分別為98.27%、98.50%、104.82%、103.08%、97.45%、98.84%、98.94%、102.79%、100.37%、103.01%,RSD分別為1.56%、1.39%、2.14%、3.68%、2.62%、1.48%、3.52%、1.28%、1.67%、1.34%,表明該方法準確度較好。
2.11 耐用性試驗
保持其他條件不變,分別考察波長、體積流量、柱溫、不同色譜柱、流動相磷酸水溶液濃度等微小變動對測定結果的影響。分別改變一維色譜條件中體積流量(0.20±0.01)mL/min、柱溫(35±2)℃、磷酸水溶液體積分數(0.050±0.005)%、不同批號色譜柱,二維色譜條件中體積流量(1.0±0.1)mL/min、柱溫(35±2)℃、磷酸水溶液體積分數(0.015±0.002)%、不同批號色譜柱,在初始條件及上述各條件下,分別進樣對照品溶液、供試品溶液,按“2.3”項下色譜條件測定,按外標法計算樣品中各成分含量。結果發現初始條件與各色譜條件變化下含量RSD均不大于5%。
2.12 樣品測定
分別取13批樣品適量,精密稱定,平行制備2份,按照“2.2”項下方法制備供試品溶液,按“2.3”項下色譜條件,按外標法計算樣品中各成分質量分數,結果見表2。JXG中測定的成分主要分為以下幾類:有機酸類包括酚酸類(原兒茶酸)和咖啡酰奎尼酸類(新綠原酸、綠原酸、隱綠原酸)、環烯醚萜苷類(馬錢苷酸、獐芽菜苦苷、龍膽苦苷、當藥苷)、糖苷類(天麻素)及嘌呤類(次黃嘌呤)。各成分含量差異較大,異構體之間存在相互轉化,因此,采用總量用于評價批次之間的差異。各成分含量與其平均含量的比值為縱坐標,化合物類型為橫坐標,作箱線圖,結果見圖3。13批制劑中天麻素、總環烯醚萜苷、總有機酸數據均分布在±30%區間內,且RSD均小于20%,表明不同批次間一致性良好。次黃嘌呤含量較低,在20~120 μg/g,各批次間差異較大,不納入批間一致性評價指標。
3 討論
3.1 色譜條件的優化
對供試品進行常規一維液相分析方法開發時,發現樣品中極性成分較多,次黃嘌呤在普通C18色譜柱上保留較弱,且出峰位置色譜峰堆積,干擾嚴重;采用DAD檢測器對天麻素、新綠原酸的光譜純度進行分析,峰純度小于990,提示可能存在包峰;原兒茶酸與相鄰組分分離度未達到1.5,不滿足分離要求;而獐芽菜苦苷在一維色譜中則易與相鄰組分共洗脫,因此,有必要對上述化合物進行第二維的分離。
為一維和二維分離選擇合適的色譜柱和流動相,是建立有效的二維色譜方法的第一步[10]。色譜柱選擇上,通常選擇顆粒填充粒徑較小、口徑較窄的色譜柱進行第一維分離,以實現良好的色譜分離度,并避免第二維色譜的峰展寬,同時可以得到較窄的峰形保證目標峰一維洗脫液不超過二維定量環體積[17-18]。
表2 13批樣品中10種成分測定結果
Table 2 Content of 10 ingredients in 13 batches of samples
批號質量分數/(mg?g?1) 次黃嘌呤天麻素原兒茶酸新綠原酸綠原酸馬錢苷酸隱綠原酸獐芽菜苦苷龍膽苦苷當藥苷 S10.0411.8540.0540.0240.2961.5420.0440.2795.4290.518 S20.1111.2240.0330.0240.2161.4810.0300.1574.9690.513 S30.0661.8030.0420.0230.2791.0520.0300.2507.3000.463 S40.0561.5750.0320.0100.2021.0690.0160.1515.8930.638 S50.0721.4960.0550.0360.3411.3080.0560.2716.2420.401 S60.0461.2710.0530.0660.2921.1210.0800.2505.4010.431 S70.0391.7180.0490.0500.2801.0970.0620.2614.7400.432 S80.0371.6170.0450.0440.2611.0580.0560.2484.5600.428 S90.0341.5760.0500.0240.2661.3600.0410.2505.1960.425 S100.0341.5960.0500.0240.2681.3780.0410.2535.2610.429 S110.0801.8930.0320.0210.2451.3900.0340.3067.1110.437 S120.0671.9720.0350.0270.2651.1290.0370.2776.2810.341 S130.0632.0780.0330.0260.2571.0950.0330.3337.3540.343
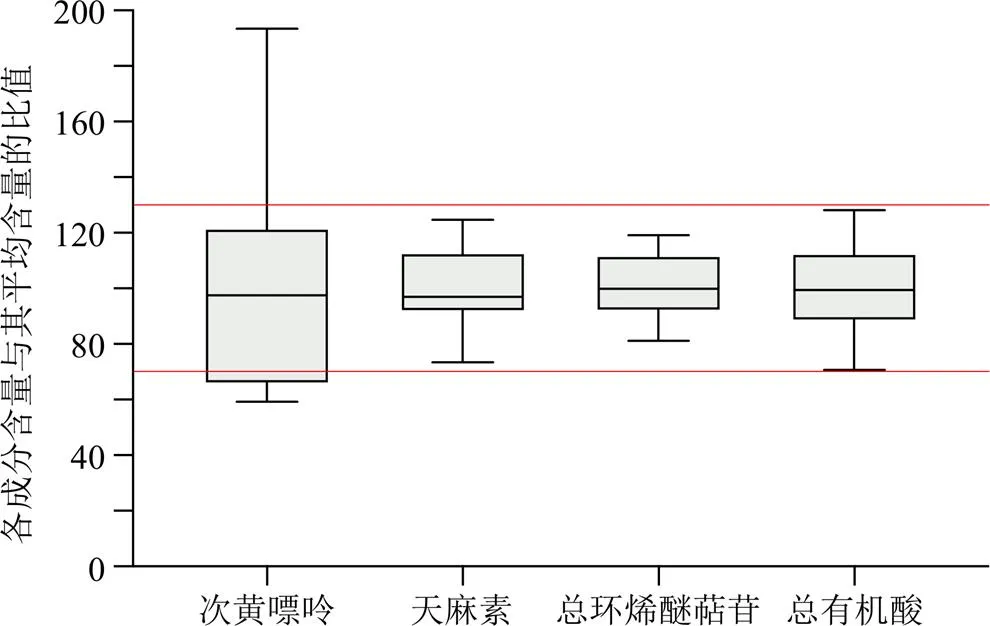
圖3 不同化合物平均含量百分比箱線圖
本研究中測試了Zorbax Eclipse Plus C18(50 mm×2.1 mm,1.8 μm)、Zorbax SB-C18(100 mm×2.1 mm,1.8 μm)以及Acquity UPLC BEH C18(100 mm×3.0 mm,1.7 μm)3種反相色譜柱,最后選擇分離效果最佳的Acquity BEH C18作為第一維色譜柱。第二維色譜柱應與第一維有一定的正交性,實驗嘗試了Cortecs C18(50 mm×4.6 mm,2.7 μm)、Xselect CSH C18(150 mm×4.6 mm,3.5 μm)、Zorbax SB-Aq(150 mm×2.1 mm,3.5 μm)、XSelect HSS T3(250 mm×4.6 mm,5 μm)等色譜柱,結果表明,XSelect HSS T3色譜柱對高比例水相流動相有較好的耐受性,且從一維洗脫的組分多屬于極性成分,在該色譜柱上有較好的分離度,因此作為第二維色譜柱。
就流動相而言,第一維采用甲醇-乙腈混合溶液和0.05%磷酸水溶液分別作為有機相和水相,為化合物提供了更適合的保留時間、更良好的分離度和峰形。實驗比較了乙腈與第一維相同溶劑作為第二維有機相的區別,發現雖然乙腈背壓會更低,且有更高的正交性,但選用與第一維相同的溶劑有更好的兼容性,可以避免出現較高的溶劑峰并保持基線平穩。在第二維水相的選擇上,由于次黃嘌呤在此系統中受pH影響較大,因此,需對水相的磷酸體積分數進行考察,比較了0.05%、0.03%、0.015%磷酸體積分數后發現,0.015%磷酸可使次黃嘌呤與相鄰組分完全分離。
3.2 供試品制備方法的選擇
實驗比較了樣品回流提取和超聲提取的提取效率,結果測得回流提取的綠原酸含量較低而新綠原酸含量較高,其他成分無明顯差別,可能是因為綠原酸及其同分異構體在高溫下進行了轉化[19-20],因此選擇超聲提取進行后續考察;考察了不同濃度的甲醇和乙醇作為溶劑的提取效果,確定50%甲醇的提取效果最佳;考察了超聲時間、溶劑用量,在考察范圍內,兩者對提取效果無明顯影響,綜合考慮確定了最終的供試品溶液制備方法。
JXG中10個成分極性與含量差異大,不同類型的色譜柱均難以達到全部分離,因此建立二維液相分析方法。本實驗基于多中心切割二維液相色譜技術建立了同時測定JXG中10種成分的分析方法,并進行了系統地方法學驗證。結果表明,該方法專屬性、穩定性、重復性、準確度均較好,測定結果穩定、可靠,可有效地反映JXG質量,二維液相的分離能力明顯優于一維液相,并可作為一維HPLC的補充方法為類似性質產品的質量控制方法提供新的思路。
利益沖突 所有作者均聲明不存在利益沖突
[1] 付娟, 李家春, 張海弢, 等. 九味熄風顆粒生物堿類成分指紋圖譜建立及其多指標成分定量分析 [J]. 中國實驗方劑學雜志, 2016, 22(19): 43-47.
[2] 杜春雁, 胡思源, 趙賓江, 等. 九味熄風顆粒治療小兒抽動障礙腎陰虧損、肝風內動證的療效觀察 [J]. 現代藥物與臨床, 2017, 32(4): 718-722.
[3] 張艷海, 張大偉, 孟兆青, 等. 在線二維液相色譜法快速測定桂枝茯苓膠囊中芍藥苷、丹皮酚、苦杏仁苷和肉桂酸的含量 [J]. 中國中藥雜志, 2013, 38(23): 4088-4093.
[4] 中國藥典[S]. 四部. 2020: 65.
[5] 張艷海, 金燕, 王崢濤. 在線二維多中心切割液相色譜法測定三七、人參及其相關產品中8種人參皂苷 [J]. 中草藥, 2017, 48(5): 894-901.
[6] Stoll D R, Carr P W. Two-dimensional liquid chromatography: A state of the art tutorial [J]., 2017, 89(1): 519-531.
[7] Qiao X, Wang Q, Song W,. A chemical profiling solution for Chinese medicine formulas using comprehensive and loop-based multiple heart-cutting two-dimensional liquid chromatography coupled with quadrupole time-of-flight mass spectrometry [J]., 2016, 1438: 198-204.
[8] Pursch M, Buckenmaier S. Loop-based multiple heart- cutting two-dimensional liquid chromatography for target analysis in complex matrices [J]., 2015, 87(10): 5310-5317.
[9] Campone L, Rizzo S, Piccinelli A L,. Determination of mycotoxins in beer by multi heart-cutting two-dimensional liquid chromatography tandem mass spectrometry method [J]., 2020, 318: 126496.
[10] Yao C L, Yang W Z, Wu W Y,. Simultaneous quantitation of fivesaponins by multi heart-cutting two-dimensional liquid chromatography: Method development and application to the quality control of eightcontaining Chinese patent medicines [J]., 2015, 1402: 71-81.
[11] Xu F, Xu Y, Liu G Z,. Separation of twelve posaconazole related stereoisomers by multiple heart- cutting chiral-chiral two-dimensional liquid chromatography [J]., 2020, 1618: 460845.
[12] Wang S Y, Zhou L N, Wang Z C,. Simultaneous metabolomics and lipidomics analysis based on novel heart-cutting two-dimensional liquid chromatography- mass spectrometry [J]., 2017, 966: 34-40.
[13] 李郭帥, 馬陽, 耿婷, 等. 超高效二維液相色譜法測定復方南星止痛膏中雙酯型烏頭生物堿的含量 [J]. 藥物分析雜志, 2019, 39(2): 249-256.
[14] 高雯, 宋慧鵬, 楊華, 等. 在線二維液相色譜技術在中藥研究中的應用進展 [J]. 色譜, 2017, 35(1): 121-128.
[15] Iguiniz M, Heinisch S. Two-dimensional liquid chromatography in pharmaceutical analysis. Instrumental aspects, trends and applications [J]., 2017, 145: 482-503.
[16] 王智聰, 傅榮杰, 吉建國, 等. 高分辨采樣二維液相色譜法同時測定金銀花中綠原酸和木犀草苷含量 [J]. 色譜, 2019, 37(2): 201-206.
[17] 張艷海, 其布勒哈斯, 金燕, 等. 在線二維液相色譜法同時測定嬰幼兒和成人配方營養品中的維生素A、D3和E [J]. 色譜, 2015, 33(3): 291-297.
[18] Qiao X, Song W, Ji S,. Separation and detection of minor constituents in herbal medicines using a combination of heart-cutting and comprehensive two- dimensional liquid chromatography [J]., 2014, 1362: 157-167.
[19] Dawidowicz A L, Typek R. Formation of ester and amine derivatives of 5--caffeoylquinic acid in the process of its simulated extraction [J]., 2012, 60(50): 12289-12295.
[20] Dawidowicz A L, Typek R. Thermal transformation of-5--caffeoylquinic acid (-5-CQA) in alcoholic solutions [J]., 2015, 167: 52-60.
Simultaneous quantitation of 10 components in Jiuwei Xifeng Granules by multiple heart-cutting two dimensional liquid chromatography
SHEN Can-jie1, 2, 3, MA Yang1, 2, CHEN Xia-Lin1, 2, GAO Xia1, 2, CAO Liang1, 2, WANG Zhen-zhong1, 2, XIAO Wei1, 2
1. Jiangsu Kanion Pharmaceutical Co., Ltd., Lianyungang 222001, China 2. State Key Laboratory of New-Tech for Chinese Medicine Pharmaceutical Process, Lianyungang 222001, China 3. China Pharmaceutical University, Nanjing 211198, China
To develop an online multiple heart-cutting two-dimensional liquid chromatography method for the simultaneous determination of 10 components from the Jiuwei Xifeng Granules (JXG).The first dimensional column was Acquity UPLC BEH C18(100 mm × 3.0 mm, 1.7 μm) and the second dimensional column was X Select HSS T3 (250 mm × 4.6 mm, 5 μm). The mobile phase was methanol-acetonitrile mixed solution (95:5) and phosphoric acid-water solution. The flow rates were 0.2 mL/min and 1.0 mL/min respectively in two dimensions. And column temperature controled at 35 ℃.The contents of hypoxanthine, gastrodin, organic acids (protocatechuic acid, neochlorogenic acid, chlorogenic acid, cryptochlorogenic acid) and iridoid glycosides (loganic acid, swertiamarine, gentiopicroside, sweroside) in JXG were determined simultaneously. Each component had a good linear relationship within the investigated concentration range (≥ 0.999), the RSD of precision were less than 2%, the RSD of repeatability were less than 5%, and the sample solution was stable within 24 h at room temperature, the average recovery were 97.45%—104.82%, and the consistency of 13 batches of test samples are good. The concentration of hypoxanthine, gastrodin, protocatechuic acid, neochlorogenic acid, chlorogenic acid, loganic acid, cryptochlorogenic acid, swertiamarine, gentiopicroside, sweroside in 13 batches of samples were 0.034—0.111, 1.224—2.078, 0.032—0.055, 0.010—0.066, 0.202—0.341, 1.052—1.542, 0.016—0.080, 0.151—0.333, 4.560—7.354, 0.341—0.638 mg/g, respectively, and the consistency of gastrodin, total organic acid and total iridoid glycosides were good.The estabilshed method is simple, accurate and reliable, which can be used for the quality control and evaluation of JXG.
Jiuwei Xifeng Granules; two-dimensional liquid chromatography; heart-cutting; simultaneous differmination; hypoxanthine; gastrodin; organic acids; protocatechuic acid; neochlorogenic acid; chlorogenic acid; cryptochlorogenic acid; iridoid glycosides; loganic acid; swertiamarine; gentiopicroside; sweroside; quality control
R286.02
A
0253 - 2670(2022)14 - 4333 - 07
10.7501/j.issn.0253-2670.2022.14.013
2021-11-30
江蘇省工信廳2020年度工業和信息產業轉型升級專項資金項目-多組分中藥研究關鍵技術
沈燦杰,碩士,研究方向為中藥分析。Tel: 18013945296 E-mail: shencanjie@163.com
肖 偉,中國工程院院士,博士,主要研究方向為中藥新藥的研究與開發。Tel: (025)86587935 E-mail: kanionxw2010@126.com
[責任編輯 鄭禮勝]