計算機模擬取樣確定標準物質的最小取樣量
2022-07-05 08:58:34江泓
中國無機分析化學 2022年3期
江 泓
(福建省地質測試研究中心,福州350001)
在化驗室內,檢測的樣品必須是均勻的。也就是說無論用什么方法從該樣品中取任何一部分子樣,對于被檢測的項目它都能代表原來的樣品。然而樣品的不均勻性是樣品的特性之一,它不能被消除,只能減小。因此絕對均勻的樣品是不存在的,只有均勻程度不同的樣品。因此,確定標準物質均勻程度,是標準物質認證的基本步驟之一。標準樣品工作導則[1]中要求在標準樣品定值過程中,同時要求進行均勻性試驗。并建議[2]標準物質證書給使用者提供使用該標準物質的最小取樣量,并要提醒使用者“只有滿足該最小樣本量要求,特性值及其不確定度才有保證。”早期的地質標準物質證書上沒有最小取樣量的信息,用戶都默認0.1 g或0.2 g為最小取樣量。進入21世紀后研制的地質標準物質以及從GS系列轉到GBW系列的早期地質標準物質基本上都提供最小取樣量的信息。但是考察這些標準物質研制報告發現,基本上都采用認證樣品均勻性時所用的樣品量為最小取樣量。實際上這是將取樣量與最小取樣量混淆了。設想一下,有一個樣品,從中取出一定量子樣做樣品均勻性檢驗,檢測結果的方差滿足標準物質定值的要求,證明樣品是均勻的;然后逐步減小子樣重量。當子樣重量減小到一定程度,檢測結果的方差不能滿足標準物質定值的要求,說明樣品不均勻。之所以會有這種現象,是因為樣品不均勻的本質造成的。無論樣品加工的粒度多么細,混勻操作多么完備,總會有少數被測物含量較高的顆粒存在。盡管這些顆粒數量很少,一旦被取到就會使結果異常偏高,檢測結果的離散程度變大。當子樣量比較大時,這個現象不太明顯。而當子樣量小時,這種現象就突出了。所以用認證均勻性時用的樣品量來代替最小取樣量是不合理的,它存在三個缺陷:第一,取樣量偏大掩蓋了樣品的不均勻性;第二,過高估計了樣品的不均勻性;第三,限制了該標準物質在微分析技術中的應用。最后一點越來越顯出它的重要性。隨著分析技術的進步的,微分析技術在實際應用中不斷普及,這些技術常只要幾十毫克、幾毫克甚至更低質量的樣品,若標準物質的最小取樣量大到幾百毫克,就不能用于監控這些技術的質量。王毅民等[3-4]在2009年就注意到這種問題,并指出“在過去標準物質研制中比較重視樣品的分析定值,而對樣品自身的特性(粒度分析、均勻性檢驗,特別是最小取樣量的確定)研究較少,相關的文獻少。”呼吁“地質分析者、標準物質研制者和實驗管理部門共同關注和解決這個問題。”建議“鼓勵支持分析方法研究者對74 μm地質標準物質的取樣量進行系統研究,進一步實驗證明最小取樣量(小于100 mg)的可行性,并公示(發表),以使最小取樣量分析的做法具有更可靠的實驗依據。”“提醒標準物質研制者……,有責任重新考察給出的最小取樣量對現代主流分析技術的適應性和確定最小取樣量方法的合理性,如與原來不符,應重新確定并發布。”然而,十幾年過去了,這些聲音并沒有得到反應。查閱最近幾年發表的標準物質研制的文獻,地質部門以外研制的標準物質仍然少有最小取樣量信息。地質部門研制的標準物質都公布了最小取樣量,但大多仍然以均勻性研究或定值時用的樣品量,通常為100 mg為最小取樣量。雖有個別低到25 mg,也不是經嚴格研究樣品的微不均勻性得來的。同時,登陸“中華標準物質網”(w.w.w.GBW365.com),GBW系列中原來GSR、GSD或GSS系列的最小取樣量沒有修訂過的跡象。另一方面,在中子活化(NAA)法或固體直接進樣塞曼石墨爐原子吸收光譜(SS-ZAAS)法用于標準物質研制和日常檢測工作中,用幾十毫克的GSS、GSD或GSR系列標準物質作質量監控的報道文獻不在少數。顯然不符合標準物質制作規范和化驗室質量管理規定。
我國標準樣品的最小取樣量確定多采用X-射線熒光光譜(XRF)法、電感耦合等離子體發射光譜(ICP-OES)法和電感耦合等離子體質譜(ICP-MS)法在均勻性檢驗時確定。PAUWELS J等[5]推薦用固體進樣塞曼石墨爐原子吸收光譜(SS-ZAAS)法和中子活化分析(NAA)法進行最小取樣量測定。這兩種方法是取樣量可小于1 mg,且不用化學預處理的分析技術之一。因為從質量小的樣品結果外推到質量大的樣品能保持樣品的分布特征。因此,用大于由此而得的最小取樣量的子樣量,能保證子樣的代表性。為了滿足統計要求,往往要求重復測定100次以上,因此配備有自動稱樣和進樣裝置的SS-ZAAS儀,分析速度快更適用。他們以二元且粒度均勻體系為基礎,推導出最小取樣量M為:
(1)

(2)
其中m為實驗取樣量,mg;cn為異常高含量顆粒對樣品含量的貢獻,%/顆;z為實驗時取到異常高含量顆粒的平均顆粒數顆;c為樣品含量值,%;HE為相對均勻常數。
(3)
則最小取樣量M:
(4)
式中,RSE為定值時的相對取樣標準偏差,%。
本文提出用計算機模擬取樣過程,對不同的取樣量重復進行測試多次,統計其檢測結果的標準偏差,作出標準偏差s對取樣量m的擬合曲線,再計算最小取樣量。計算機運算速度快,參數轉換方便,也可以取毫克甚至小于毫克數量級的樣進行實驗。設計的模式是一顆一顆地取樣,這樣不存在取樣操作及測試過程帶來的誤差。只要有足夠的巖礦鑒定的資料,可以模擬不同狀態下的樣品取樣過程。
1 實驗部分
1.1 軟件
實驗程序用Visual Basic軟件編寫和運行。
1.2 樣品
沒有標準樣品,只能采用模擬的樣品。模擬黃銅礦樣品,主要成分為黃銅礦,其余礦物都歸到脈石中去的二元體系。ω(Cu)=0.230%±0.005%,黃銅礦中ω(Cu)=34.56%,脈石中ω(Cu)=0.03%,銅樣品的粒度分布如表1所示。

表1 模擬標準物質粒度分布[7]
黃銅礦密度為4.45 g/cm3,脈石的密度為2.75 g/cm3。黃銅礦與脈石完全解離。顆粒形狀均為立方體。根據這些參數同時考慮到顆粒小于1 μm時,單顆粒對檢測結果影響極小,而且比例不大,將它們的重量歸并到1.04 μm粒級中。由此計算出200 g樣品中各粒級的顆粒數和總顆粒數(表2)。

表2 200 g模擬樣品各粒級的顆粒數
黃銅礦和脈石各分成7個不同粒徑段,共14個樣段。每一段內顆粒粒徑、形狀和銅含量等參數都一樣。
1.3 模擬取樣
1)將200 g樣的13734306964585粒樣品,按黃銅礦7個段,再按脈石7個段,由粗到細逐個編號,每粒一號,不重復。
2)將各段的顆粒數相加即200 g樣的總顆粒數,構成一個隨機數池。計算機用Visual Basic軟件的隨機函數從隨機池中取一個數,代表取一粒樣品。
3)每取一粒樣品都檢查它是哪一段的。則該段的被取顆粒數加1;被取的顆粒數乘以該段的單顆粒重,即為該段被取的樣重;同時該段的顆粒數減1。
4)每取一粒樣品后都要將各段被取的樣重加和求得此時被取的子樣重量。
5)將此時被取的子樣重與預設的子樣重比較。若被取的子樣重小于預設的子樣重,則重復2)到5)步驟,直到被取子樣重比預設子樣重。
6)計算被取子樣中銅的含量:
(5)
其中,mineral是子樣中黃銅礦重,是黃銅礦顆粒數與單顆黃銅礦重之積,g;gangue是子樣中脈石重;是單顆脈石重與脈石顆粒數之積,g;W為被取子樣重,g。
7)重復1)-6)步驟30次。計算30次銅含量結果的標準偏差,程序見圖1。

圖1 模擬取樣程序框圖Figure 1 The flow chart of simulating program.
2 結果與討論
模擬30次取不同子樣實驗結果如表3所示,其中m為子樣重;c為平均含量;s為標準偏差;n為子樣中最大粒徑(40.74 μm)黃銅礦的平均值顆粒數。
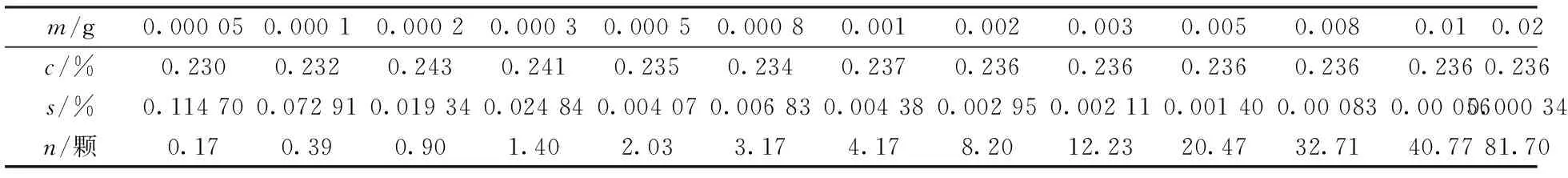
表3 模擬取樣實驗結果
2.1 取樣量與誤差的關系
根據表3作出取樣量與誤差的關系見圖2。圖2中m-s的擬合曲線,橫坐標為取樣量m(g),左邊縱坐標為標準偏差s(%),右邊縱坐標為子樣中平均mineral顆粒數(顆)。擬合結果為冪函數,相關系數為0.963 9。
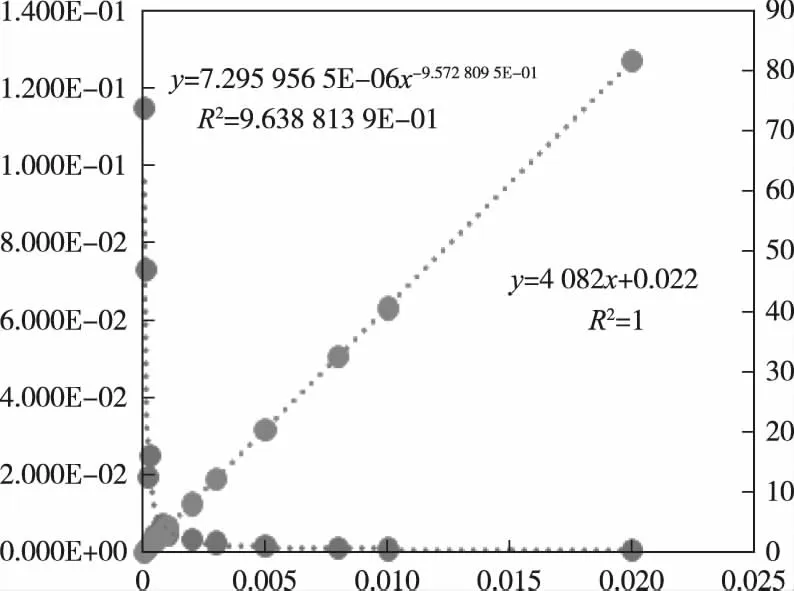
圖2 m-s及m-n關系圖Figure 2 The reltions of m-s and m-n.
s=0.000007296m-0.9573
(6)

2.2 取樣量m與取得含量銅量異常高顆粒數n的關系
本實驗用的樣品最大的顆粒徑為40.74 μm,表3第4行為子樣中粒徑為40.74 μm黃銅礦30次實驗的平均值。子樣量與銅量異常高顆粒數關系見圖2。擬合結果為直線,相關系數為r=1,解析式為:
n=4082.6m+0.0223
(7)
數理統計認為當異常高顆粒平均顆粒數n≥9時,體系就從泊松分布轉變成正態分布。因此將9代入(7)式:
兩種方法得到結果比較接近,應當取4.1 mg為該黃銅礦樣的最小取樣量。
INGAMELLS[8-10]推導出的取樣常數方程為:
s=Ksm-1
(8)
在前階段[11]的計算機模擬取樣確定取樣常數研究中,也證實對于粒度均勻體系,這個公式是成立的。但從式(6)可見m-s的關系式與(8)式形式一樣都是冪函數,指數卻不同。原因可能是由于INGAMELLS推導時用的顆粒均勻的二元體系,而本模擬實驗用的是有一定粒度分布的二元體系所致。式(6)與PIERRE M GY的取樣理論也不同。KRATOCHVIL等[12-13]認為粒度分布不影響子樣量m的指數,只影響不均勻常數Ks中的粒度分布指數,粒度分布指數隨著粒度分布變寬從1逐漸變小。從實驗過程觀察推測s-m關系式推測應當是:
s=Ksm-n
(9)
n為0.5~1,從均勻粒度隨粒度分布越來越廣而增大,同時Ks也會變化。這一點以后還要設計實驗來考察。
3 結論
1)計算機模擬黃銅礦樣品取樣,取xmg甚至0.xmg的子樣研究樣品的不均勻性,它不受取樣操作和檢測過程誤差的干擾,誤差只來自樣品本身的不均勻性,操作是可行的。通過s-m擬合曲線和定值時的樣品不均勻誤差計算最小取樣量,結果是可靠的。計算機還具有運算速度快,參數變換方便等優點。SS-ZAAS法和NAA法雖然設備比較貴,保養條件要求高,現在不少單位也開始配備,標準物質研制單位也應當充分利用它開展標準物質的最小取樣量研究。
2)模擬的黃銅礦樣品最小取樣量為4.1 mg,假設它是標準樣品并用它來監控SS-ZAAS法等微分析技術檢測,進樣量要大于4.1 mg才可使檢測結果受控。若要進樣0.xmg檢測必須改用最小取樣量更小的標準樣品。從標準樣品的研制實踐看,現在例行的粉碎混勻設備與流程大部分能保證-0.074 mm粒度達99%以上,最大粒徑大都在0.040~0.060 mm。最小取樣量基本上都在xmg水平。若要制得最小取樣量達0.xmg級,氣流粉碎是可值得研究的方向。
3)對于s-m的擬合結果函數關系式,還要進一步設計實驗研究。
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
當代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
專用汽車(2016年4期)2016-03-01 04:13:43
