電感耦合等離子體原子發射光譜(ICP-AES)法測定高鎳銅液體樣品中金、鉑、鈀
2022-07-05 08:58:52段愛霞李希凱祖愛國趙志虎李立香段正輝楊秀萍楊紅玉
中國無機分析化學 2022年3期
關鍵詞:實驗
段愛霞 李希凱* 趙 勇 祖愛國 趙志虎 李立香 段正輝 楊秀萍 楊紅玉
(1.金川集團股份有限公司 檢測中心,甘肅 金昌 737100;2.金川集團股份有限公司 鎳鈷研究設計院,甘肅 金昌 737100;3.金川集團股份有限公司 鎳冶煉廠,甘肅 金昌 737100)
近年來,國內礦產資源的需求呈大幅增長的趨勢,但在礦產資源的選冶和綜合利用過程中存在部分元素回收率低的問題。故提升礦產資源的回收率非常必要,尤其是鎳銅冶煉的中間物料[1]。如:貴浸液、銀硒液、1.3萬t加壓浸出液、二鎳氯氣浸出原液等中間物料。這些樣品組分復雜,除了含有大量的鎳(最高250 g/L)、銅(最高50 g/L)基體,還含有少量的鈷、鉻、鉛、銀等其他元素,而稀貴金屬金鈀鉑的含量(0.01~0.57 g/t)甚微,缺乏具有針對性的測定方法。
對于部分樣品,其中還含有難以溶解的沉淀物,樣品的均勻性較差。相對于大量基體,對含量極低的金鉑鈀要實現同時直接測定幾乎不可能,主要原因是大量基體的干擾和共存元素的干擾問題,因此,必須降低大量基體的共存量,并對金、鉑、鈀元素進行富集,才能實現上述檢測目的。本文通過碲共沉淀,貴金屬進入濾渣,經火試金分離富集得到貴金屬合粒,合粒經王水溶解后,利用電感耦合等離子體光譜儀靈敏度高、檢出限低、穩定性好、干擾少、線性范圍寬、檢測速度快、能實現多元素同時測定等優點[2-6],在電感耦合等離子體光譜儀上同時測定金、鈀、鉑的量,為生產工藝提供了準確、快捷的分析數據。
1 實驗部分
1.1 主要儀器與試劑
高溫箱式電阻爐(最高使用溫度1 350 ℃),ICAP 7400電感耦合等離子體光譜儀(賽默飛世爾科技公司),黏土坩堝(材料為耐火黏土,4#),鎂砂灰皿,鑄鐵模。
氧化鉛(粉狀,wAu<0.05 g/t,wPt<0.05 g/t,wPd<0.05 g/t,試金級),無水碳酸鈉(粉狀,工業純),石英砂(粉狀,分析純),硼砂(粉狀,工業純),面粉,硝酸鉀(粉狀,分析純),硝酸銀溶液(63 g/L),硝酸(ρ1.42 g/mL),鹽酸(ρ1.19 g/mL),硝酸(1+1),鹽酸(2+1),過氧化氫(30%),氯化鈉(200 g/L),王水(現配現用),氯化亞錫溶液[50 g SnCl2.2H2O溶于100 mL HCl(1+1)中],四氯化碲溶液[碲(1 g/L):取100 mg碲,加2 mL硝酸,蒸干,加1 mL鹽酸,再蒸干,用10 mL濃鹽酸溶解殘渣,并用水稀釋至100 mL],硫酸肼溶液(50 g/L),金標準儲備溶液(l g/L),鈀標準儲備溶液(l g/L),鉑標準儲備溶液(l g/L),金、鈀、鉑混合標準溶液(50 μg/mL),金、鈀、鉑混合標準溶液(5 μg/mL)。
1.2 實驗方法
移取25 mL樣品于50 mL預先放有少許濾紙漿的比色管中,加入10 mL鹽酸、4 mL碲溶液,混勻,放置10 min,加入3 mL硫酸肼溶液(50 g/L),不斷攪拌下滴加5 mL氯化亞錫溶液,于60~70 ℃水浴中加熱30 min。過濾,棄去濾液,濾紙及殘渣放入坩堝中,加入1.0 g面粉、30 g碳酸鈉、8 g硼砂、150 g氧化鉛、10 g二氧化硅,置于900 ℃的試金爐中,關閉爐門,升溫至1 160 ℃,保溫10 min后出爐。將鉛扣灰吹。所得合粒加入5 mL硝酸,加熱溶解至淡黃色煙冒盡溶液無色時取下,加入20 mL鹽酸,蒸發至干,加入1 mL王水,加熱到沸騰,用水沖洗表皿及杯壁控制體積20 mL左右,煮沸至溶液澄清,用水移入25 ml比色管中并定容。在儀器選定的最佳工作條件下用ICP-AES法測定金、鈀、鉑的含量。
1.3 空白實驗
隨同試料做氧化鉛空白實驗。測定方法與樣品相同。
1.4 標準曲線的繪制
移取0、1.00、2.00、4.00、10.00 mL金、鈀、鉑混合標準溶液分別置于一組100 mL容量瓶中,加入5 mL王水,用水定容,混勻。在選定的儀器條件下,于電感耦合等離子體光譜儀上測定金、鉑、鈀各元素的強度,儀器自動繪制金、鉑、鈀的工作曲線。
1.5 金、鉑、鈀量的測定
繪好工作曲線后,于電感耦合等離子體光譜儀上,測定試液中金、鉑、鈀的發射強度,根據工作曲線得到試樣中金、鉑、鈀的含量。按式(1)計算金、鉑、鈀的含量,數值以μg/mL表示。
(1)
式中:
ρx——被測試液中金、鉑、鈀的濃度,μg/mL;
ρ——自工作曲線上查得的金(鈀、鉑)量濃度,μg/mL;
V0——測定時試液的體積,mL;
V——分取試液體積,mL;
計算結果表示至小數點后兩位。
2 結果與討論
2.1 ICP-AES儀器測定條件
從ICP-AES的射頻發生器功率、霧化氣流量、等離子體氣流量、進液泵速和儀器推薦條件等方面對被測元素譜線發射強度考慮,最終選擇的儀器測定參數見表1。

表1 ICP-AES儀器工作參數
2.2 各元素分析譜線的選擇
遵循所選譜線靈敏度高,干擾少的原則,在同一條件下,同時測定每種元素各譜線的強度及其周圍的干擾譜線,確定金、鉑、鈀元素的分析譜線分別是:267.595、265.945和340.458 nm。
2.3 檢出限及測定下限
試劑空白連續11次測定,計算其標準偏差,3倍的標準偏差所對應的濃度為本方法的檢出限,10倍的標準偏差為測定下限。數據見表2。
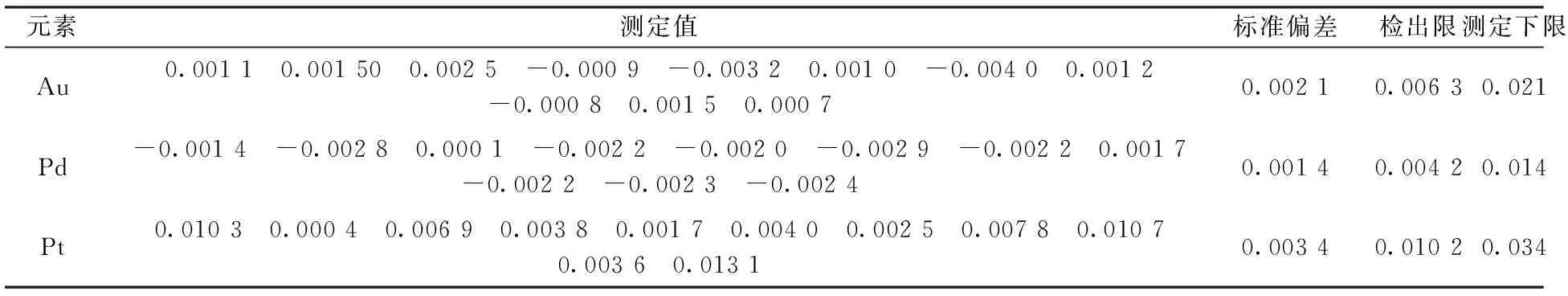
表2 方法的檢出限和測定下限
從表2數據可知,各元素的測定下限能滿足高鎳、銅液體樣品中金、鉑、鈀的測定要求。
2.4 樣品分離富集方法的選擇
2.4.1 碲共沉淀富集酸度的選擇
取含20 μg金鈀鉑混合標準溶液于一組100 mL燒杯中,調整鹽酸濃度為1.0、2.0、3.0、4.0、5.0 mol/L,控制溶液總體積20 mL,按實驗方法進行共沉淀分離回收率實驗,結果見表3。
表3結果表明,1~5 mol/L鹽酸濃度,均能定量共沉淀,考慮到低酸度條件下,氯化亞錫被氧化后容易水解,過濾較慢,實驗選擇鹽酸濃度為1.5~3 mol/L。
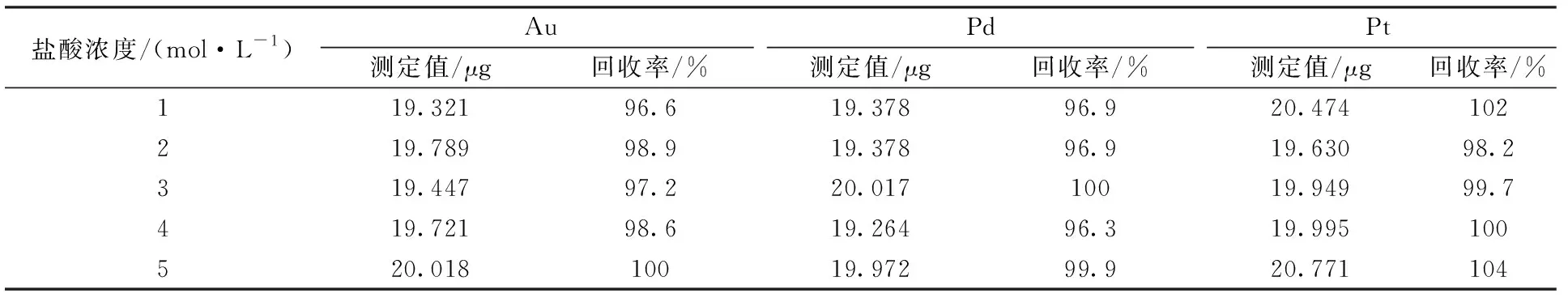
表3 鹽酸加入量對共沉淀的影響
2.4.2 碲加入量選擇
為保證加入的碲量與溶液中Au、Pt、Pd定量共沉淀而與雜質元素分離,分別取20 μg金鈀鉑混合標準溶液于一組100 mL燒杯中,調鹽酸濃度3.0 mol/L,分別加入2.0、3.0、4.0、5.0 mg碲,按實驗步驟共沉淀分離后做回收率實驗,結果見表4。
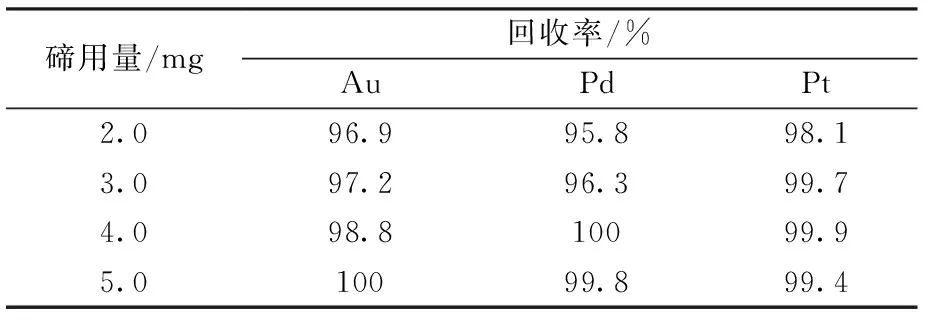
表4 碲加入量對共沉淀的影響
當加入2.0~5.0 mg的碲時,回收率在95.8%~100%,說明均能沉淀完全。考慮到高基體可能會影響沉淀效果,以及加入過多碲時會夾帶較大的銅和鐵等雜質,實驗選擇加入的碲量為4.0 mg。
2.5 火試金配料方案的確定
貴浸液、銀硒液、1.3萬t加壓浸出液、二鎳氯氣浸出原液等中間物料,主要含鎳、銅、鐵、鈷,含量范圍見表5。
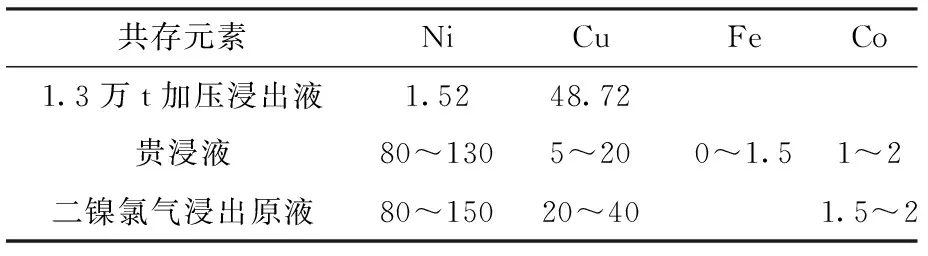
表5 樣品中主要的共存元素及其含量范圍
樣品中鎳、銅的化合狀態較復雜,按照含鎳銅樣品的配料要求,火試金熔煉后熔渣的硅酸度一般控制在1.0以下,硅酸度太高易生成冰鎳或冰銅,造成貴金屬損失[1],氧化鎳在氧化鉛中溶解度很小,在熔煉中難以和二氧化硅形成鎳的硅酸鹽進入渣中,為避免鎳進入鉛扣中,通常采用加大氧化鉛用量和加入較多的硼砂代替一部分二氧化硅,使酸性更強的三氧化硼與本來難以和二氧化硅生成硅酸鹽的氧化鎳、氧化鐵生成相應的硼酸鹽。
根據硝石法配料的原則和樣品中各成分的含量范圍,選用1個樣品,采用不同硅酸度的熔劑進行實驗,得到重量為30~40 g鉛扣時的配料及實驗現象見表6。
從表6可知,硅酸度過高,配料為半熔態,鉛粒不能聚集,硅酸度過低,熔渣成強堿性,對坩堝腐蝕嚴重。最終,選擇熔渣流動性較好的0.75硅酸度相應的配料比。

表6 不同硅酸度實驗
2.6 熔煉溫度的選擇
熔煉溫度過高,突然反應產生的氣體會使物料濺出,熔煉溫度過低,會使熔渣與鉛扣分離不徹底,試金失敗。故最終熔煉溫度為:試樣在900 ℃下進爐,升溫至1 160 ℃后,保溫約10 min出爐。
3 樣品分析
3.1 精密度實驗
選取3個代表性試樣,按照實驗步驟,對3個樣品測定11次,計算相對標準偏差。結果見表7。
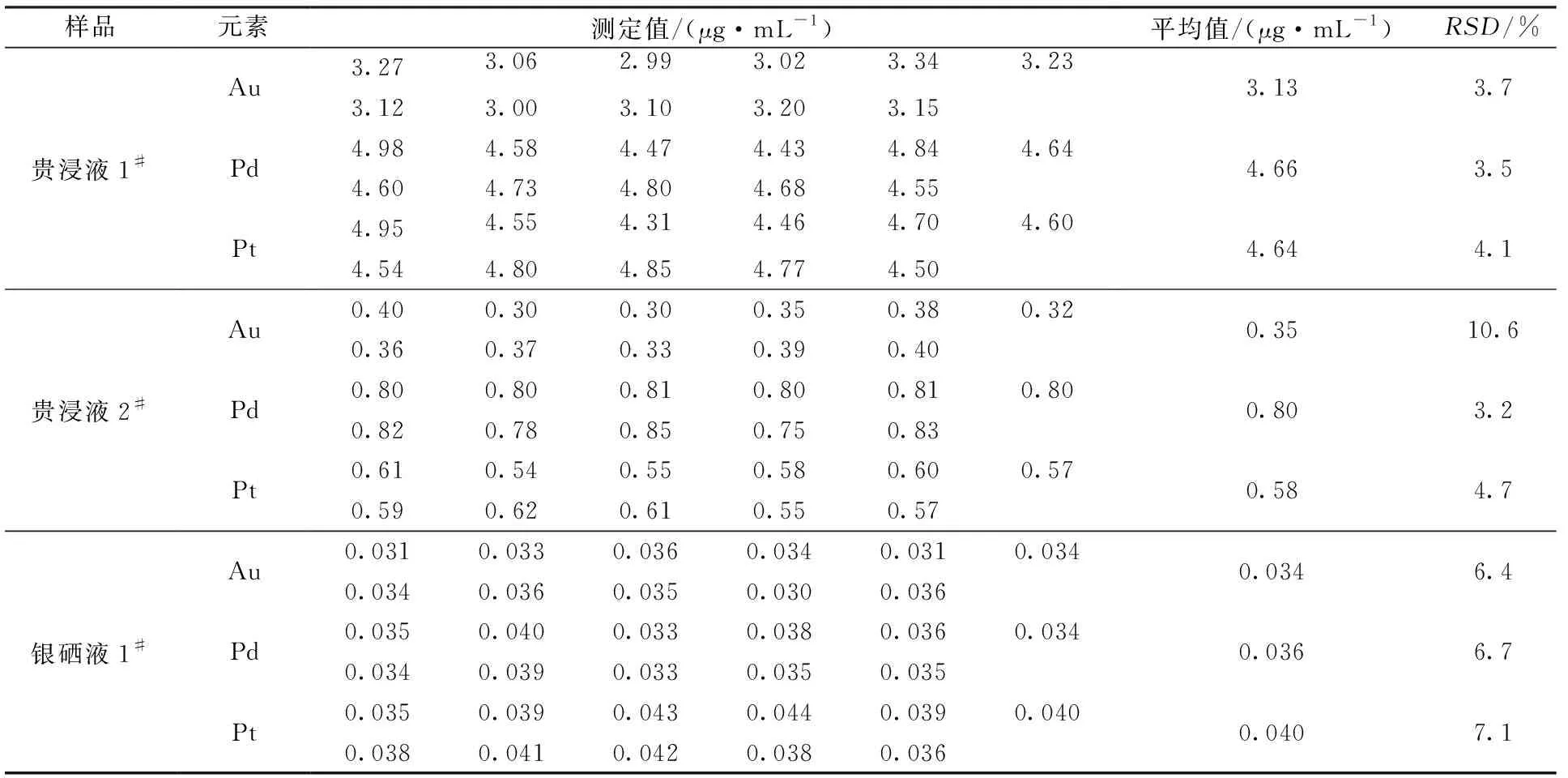
表7 方法的精密度
從表7數據中可以看出,高鎳、銅液體樣品中3種元素的RSD為3.2%~10.6%,可以滿足高鎳銅液體樣品中金、鉑、鈀元素的測定需求。
3.2 加標回收實驗
選擇2批具有代表性的樣品銀硒液和貴浸液,加入5 μg/mL的混合標準溶液,按實驗步驟,在選定儀器條件下進行加標實驗,測得的數據見表8。
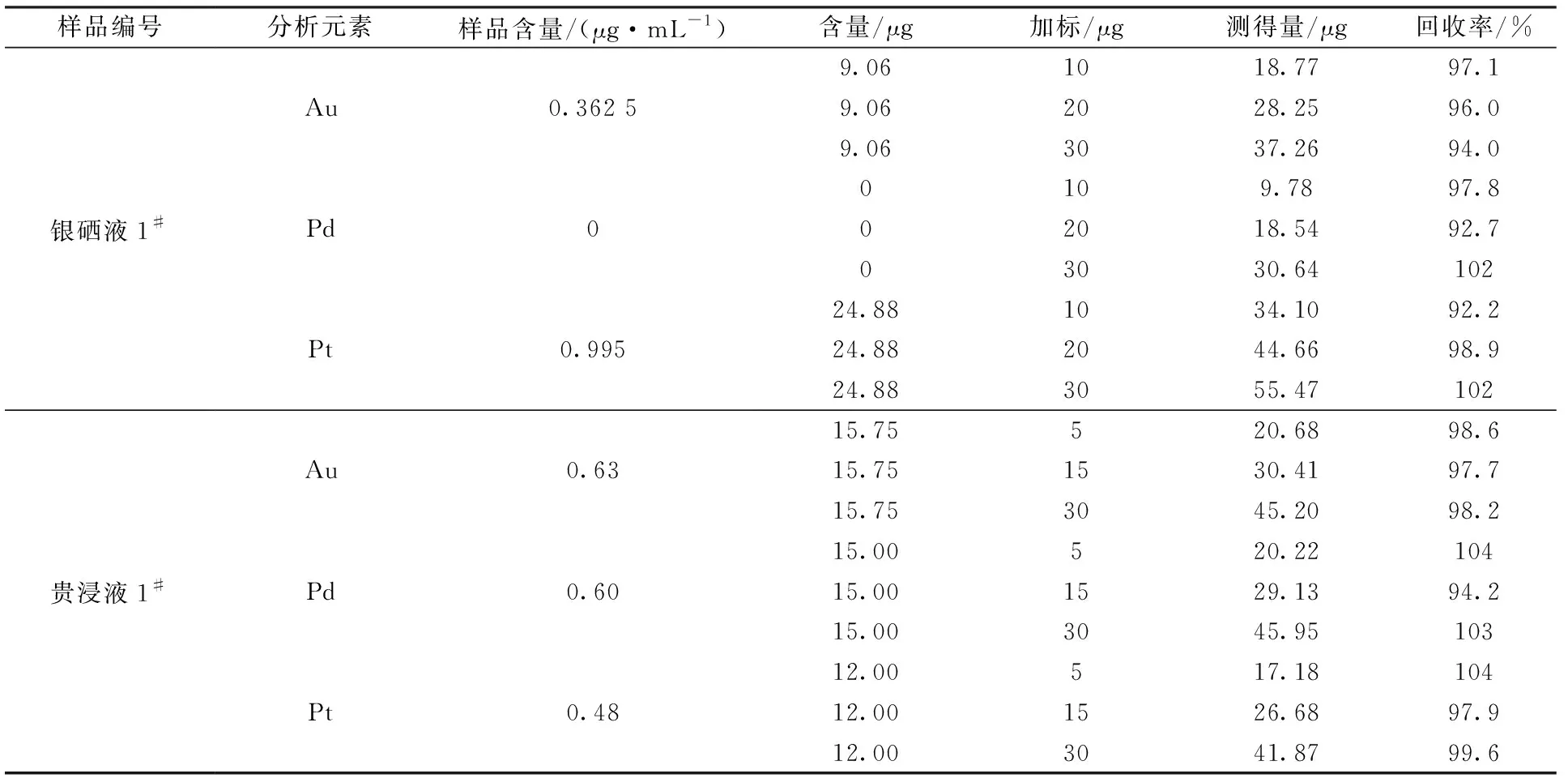
表8 加標回收實驗
結果表明,金、鉑、鈀各元素的加標回收率在92.2%~104%,結果滿意,說明碲共沉淀-火試金分離富集處理高鎳、銅溶液樣品可行。
4 結論
通過碲共沉淀和火試金技術,消除了鎳、銅干擾,實現了高基體液體樣品中低含量金鈀鉑的ICP-ASE測定。方法精密度好,準確度高,完全能夠滿足鎳、銅冶煉中間控制分析的需要。該技術對高鎳、銅基體樣品中低含量金、鉑、鈀各元素的測定有推廣價值。
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
