3種食源性致病菌 TaqMan 多重熒光定量PCR檢測方法的建立
2022-04-24 02:51:58王華健趙志強趙興華李杰峰
畜牧獸醫學報 2022年4期
王華健,張 寧,楊 威,趙志強,李 茜,陸 安,田 勇,何 欣,趙興華,4*,李杰峰*
(1.河北農業大學動物醫學院,保定 071000;2.河北省畜牧獸醫研究所,保定 071000;3.石家莊市金元康牧藥業有限公司,石家莊 051130;4.河北省獸醫生物技術創新中心,保定 071000)
食源性疾病是食品安全的主要問題之一,據世界衛生組織(World Health Organization, WHO)統計報告,全球平均每年有上億腹瀉病例,以兒童死亡率最高,其中超70%疾病由致病微生物所致[1]。自2015年我國系統開展食源性疾病監測以來,新的食源性細菌感染不斷出現,大腸桿菌O157∶H7和產單核細胞李氏桿菌感染是近年重要的食源性疾病[2]。大腸桿菌O157∶H7是腸出血性大腸桿菌(enterohemorrhagicEscherichiacoli, EHEC)的代表血清型,1999年、2000年相繼在我國安徽和江蘇兩省暴發流行,并波及中原及西部地區,在華北、東北及華東少數地區也出現了散發病例。沙門菌在我國分布廣泛,劉繼開等[3]調查研究發現除西藏外所有省份均有檢出,最常見血清型為鼠傷寒型和腸炎型,除此之外,近年來,非傷寒型沙門菌(NTS)也被認為是我國食源性疾病的主要病原菌。WHO將產單核細胞李氏桿菌列為與高住院率和死亡率有關的主要食源性病原體。2015年,美國疾病預防控制中心(CDC)報道了一起由產單核細胞李氏桿菌引起的食源性疾病暴發事件,至少8人因食用藍鈴(Bule Bell)公司冰淇淋產品患病,導致3人死亡。國外研究發現生鮮肉中產單核細胞李氏桿菌污染最嚴重,對四環素類、氯霉素和青霉素等抗生素耐藥[4-5],國內牛桓彩等[6]通過對產單核細胞李氏桿菌流行病學和耐藥性研究也有相似結論。綜上,大腸桿菌O157∶H7、沙門菌和產單核細胞李氏桿菌在食品中污染嚴重、危害巨大,針對這3種食源性致病菌建立一種快速檢測方法非常必要。
食源性致病菌檢測方法中,目前最常用的是細菌分離培養結合生化鑒定,但該傳統方法費時、費力且不能對食源性致病菌進行深入分析。分子生物學經過幾十年的發展,已經廣泛應用于食源性致病菌的檢測,基于核酸和熒光標記的熒光定量PCR(qPCR)方法作為先進的技術手段,具有特異性強、靈敏度高、耗時短等優點[7]。目前,應用qPCR方法針對一種或多種食源性致病菌的檢測穩定,但未見關于這3種致病菌的多重qPCR檢測方法的研究和市場專利產品等。rfbE基因是大腸桿菌O157∶H7的特異基因,rfb基因簇編碼多種將低聚糖單元組裝成特異O抗原所必需的酶[8];沙門菌吸附和侵襲上皮細胞表面蛋白由invA基因合成,是主要毒力因子且核苷酸序列保守[9];產單核細胞李氏桿菌溶血素編碼基因hlyA高度保守和特異[10]。本研究基于3種致病菌保守性基因擬建立一種可同時檢測這3種食源性致病菌的多重qPCR方法,用于食品中致病菌污染檢測。
1 材料與方法
1.1 標準菌株
試驗所用標準菌株共10株,目標菌株為大腸桿菌O157∶H7(EscherichiacoliO157∶H7,NCTC 12900)、沙門菌(Salmonela)和產單核細胞李氏桿菌(Listeriamonocytogenes)3種,其余7種供試菌株有大腸桿菌(Escherichiacoli)、表皮葡萄球菌(Staphylococcusepidermidis)、金黃色葡萄球菌(Staphylococcusaureus)、克雷伯菌(Klebsiellaspp.)、巴氏桿菌(Pasteurella)、豬鏈球菌2型(Streptococcussuistype 2)和糞腸球菌(Enterococcusfaecalis),所有菌株均保存于河北省畜牧獸醫研究所微生物室菌種保藏庫。
1.2 主要試劑和培養基
通用型DNA提取試劑盒、M5 GoldStar TaqMan Mixture(北京聚合美生物科技有限公司);溶菌酶(北京索萊寶科技有限公司);營養肉湯(nutrient broth, NB)、改良EC肉湯(Modified EC Broth, mEC+n)、改良山梨醇麥康凱瓊脂(Modified Sorbitol MacConkey Agar, CT-SMAC)、緩沖蛋白胨水(buffered peptone water, BPW)、四硫磺酸鹽煌綠增菌液基礎(tetrathionate broth base, TTB)、亞硒酸鹽胱氨酸增菌液(elenite cystine broth, SC)、沙門氏菌顯色培養基(第二代)、李氏菌增菌肉湯基礎(Listeriaenrichment broth base, LB1, LB2)、PALCAM 瓊脂(青島海博生物技術有限公司);法國科瑪嘉李斯特菌顯色培養基(上海欣中生物工程有限公司)。
1.3 主要儀器
Light Cycler?96實時熒光定量PCR儀(德國羅氏診斷有限公司)、K5500超微量分光光度計(北京凱奧科技發展有限公司)。
1.4 引物和探針
登錄NCBI網站,在GenBank中分別下載大腸桿菌O157∶H7、沙門菌和產單核細胞李氏桿菌的保守基因rfbE、invA和hlyA序列。應用MegAlign軟件進行序列比對,選擇合適的基因序列,應用Primer Express 3.0.1軟件設計引物和TaqMan探針。引物和探針序列見表1所示,各引物和探針均由上海生工生物工程股份有限公司合成。

表1 引物和探針序列
1.5 菌株培養及細菌基因組DNA提取
將大腸桿菌O157∶H7、沙門菌和產單核細胞李氏桿菌分別接種于NB中復蘇,37 ℃ 培養12 h后進行平板培養基分離[大腸桿菌O157∶H7,改良山梨醇麥康凱瓊脂(CT-SMAC)培養基;沙門菌,沙門氏菌顯色培養基(第二代);產單核細胞李氏桿菌,PALCAM 瓊脂培養基]。在各平板上挑取單個菌落于NB中37 ℃ 純培養12 h,取1 mL細菌純培養液,按通用型DNA提取試劑盒提取各細菌基因組DNA。用K5500超微量分光光度計測定各細菌DNA模板的濃度和純度,并計算DNA拷貝數。DNA模板于-20 ℃保存備用。
1.6 單重qPCR 擴增及特異性試驗
分別以目標菌株和供試菌株的基因組DNA為模板進行擴增。反應體系:2×M5 GoldStar TaqMan Mixture 10 μL,上下游引物各0.6 μL,探針0.4 μL,DNA模板2 μL,ddH2O補足至20 μL。引物和探針濃度為10 mmol·μL-1。反應參數:95 ℃預變性15 min;95 ℃變性3 s,60 ℃退火并延伸30 s,40個循環,在每個循環的退火延伸階段收集熒光信號。以NB培養液為陰性對照,無菌超純水為空白對照。
1.7 多重qPCR 體系的建立和優化
以單重qPCR反應體系為基礎,對3株目標菌株引物和探針的使用量進行優化,確認最優多重qPCR反應體系。
1.7.1 多重qPCR特異性試驗和標準曲線的建立 同步驟“1.5”提取各目標菌株的DNA模板并用ddH2O倍比稀釋為濃度107~103copies·μL-1,將3種目標菌株的DNA模板按每個稀釋度等量混勻并做3個重復,按優化后的qPCR反應體系,分別對大腸桿菌O157∶H7、沙門菌、產單核細胞李氏桿菌和其他7株供試菌種進行多重qPCR擴增,構建標準曲線。
1.7.2 多重qPCR靈敏性和重復性試驗 同步驟“1.7.1”,將3種目標菌株的DNA模板等量混勻,倍比稀釋濃度為107~100copies·μL-1,按優化后的反應條件進行多重qPCR擴增確定其靈敏性,每個稀釋度設3個重復。重復性試驗根據靈敏性試驗選擇合適的DNA模板濃度,每隔5 d做一組重復性試驗,共3次。根據結果分別計算批內變異系數(CV%)和批間變異系數。
1.8 臨床樣品檢測
在本地超市、農貿市場和商店中購買120份生鮮肉,包括雞肉41份、豬肉47份、牛肉18份、羊肉14份。以進出口行業標準(SN/T 1870—2016《出口食品中食源性致病菌檢測方法實時熒光PCR法》)為基礎,按優化后的多重qPCR反應體系進行樣品檢測,同時根據國家標準(GB 4789.36—2016《食品安全國家標準食品微生物學檢驗 大腸埃希氏菌O157∶H7/NM檢驗》、GB 4789.4—2016《食品安全國家標準 食品微生物學檢驗 沙門氏菌檢驗》、GB 4789.30—2016《食品安全國家標準 食品微生物學檢驗 單核細胞增生李斯特氏菌檢驗》)進行檢測結果對比,驗證多重qPCR方法的準確性。
1.9 數據統計
應用SPSS 21.0軟件對試驗數據進行統計分析。
2 結 果
2.1 DNA模板濃度和純度的測定及單重qPCR特異性
DNA模板濃度和純度(A260nm/A280nm)測定數值如下:大腸桿菌O157∶H7分別為166.39 ng·μL-1和1.799,沙門菌分別為272.62 ng·μL-1和1.847,產單核細胞李氏桿菌分別為35.905 ng·μL-1和1.967。拷貝數計算公式:DNA拷貝數=[6.02×1023(copies·mol-1)×DNA濃度(g·μL-1)]/目的基因組的分子量,式中需要的條件有DNA濃度和目的基因組的分子量。沙門菌基因組大小為4.8 Mb,1 bp堿基分子量為652 g·mol-1[11];大腸桿菌O157∶H7基因組大小約4.2 Mb[12];產單核細胞李氏桿菌基因組大小約3 Mb[13]。3種目標菌株分別進行單重qPCR反應,結果見圖1所示,擴增曲線明顯,其他供試菌株均無擴增曲線,表明試驗設計的3種目標菌株的引物和探針特異性良好。

1.大腸桿菌O157∶H7;2.沙門菌;3.產單核細胞李氏桿菌;4~11.其他供試菌株(E. coli、S. epidermidis、S. aureus、Klebsiella、Pasteurella、S. suis type 2、E. faecalis)和空白對照
2.2 多重qPCR反應體系的優化
以引物和探針濃度為10 mmol·μL-1為基礎,對多重 qPCR 反應體系的各引物、探針和Mixture等用量進行優化,選出最優反應體系為2×M5 GoldStar TaqMan Mixture 30 μL,3種細菌混合上、下游引物1.5 μL,混合探針0.6 μL,混合模板4 μL,ddH2O 補足至50 μL。反應參數:95 ℃預變性15 min;95 ℃變性3 s,60 ℃退火并延伸30 s,40個循環,在每個循環的退火延伸階段收集熒光信號。
2.3 多重qPCR特異性和標準曲線的建立
多重qPCR特異性試驗結果為3種目標細菌均顯示良好的擴增曲線,非目標細菌和空白對照均無擴增,表明優化后的多重qPCR反應對大腸桿菌O157∶H7、沙門菌和產單核細胞李氏桿菌具有良好的特異性,且不產生交叉反應。多重qPCR標準曲線結果見圖2,大腸桿菌O157∶H7的標準曲線線性方程為y=-3.743x+45.675,相關系數R2= 0.999,擴增效率為85%;沙門菌的標準曲線線性方程為y=-3.62x+43.13,R2=0.999,擴增效率為88.9%;產單核細胞李氏桿菌的標準曲線線性方程為y=-3.926x+44.296,R2=0.995,擴增效率為80%。R2均大于0.99,表明DNA拷貝數濃度的對數值(X軸)與Ct值(Y軸)之間具有良好的線性關系,引物和探針的擴增效率較高,建立的多重 qPCR 反應體系穩定。

1.大腸桿菌O157∶H7;2.沙門菌;3.產單核細胞李氏桿菌
2.4 多重qPCR靈敏性和重復性
多重qPCR靈敏性試驗陽性結果判定為Ct≤35[14-15],qPCR試驗通常設定為40個循環,當Ct值>35時,反應的隨機性增大,重復性試驗誤差增加,可能伴隨非特異性產物的影響。大腸桿菌O157∶H7、沙門菌和產單核細胞李氏桿菌的靈敏性均為104copies·μL-1。根據重復性試驗結果選擇濃度梯度為107~104copies·μL-1的DNA模板,4個梯度批內變異系數為0.09%~2.97%,批間變異系數為0.23%~7.82%。變異系數只在平均值不為零時有意義,反應單位均值上的離散程度,當批內變異系數小于5%~10%,批間變異系數小于15%~20%時,表明試驗結果可重復性高,建立的多重qPCR檢測方法重復性好,具體數據見表2和表3。

表2 多重qPCR批內重復性試驗
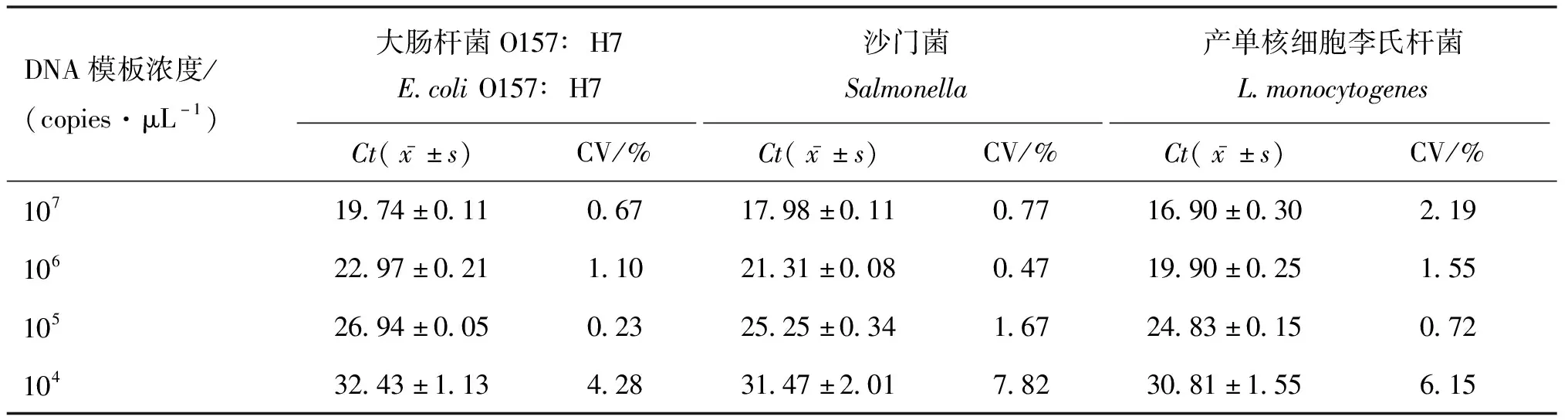
表3 多重qPCR批間重復性試驗
2.5 臨床樣品檢測
以國家標準檢測方法為對照,檢驗已建立的多重 qPCR方法的準確性,結果對比見表4。市場采集的120份生鮮肉樣品中,國家標準方法檢測出2份大腸桿菌O157∶H7、13份沙門菌和21份產單核細胞李氏桿菌;使用建立的多重qPCR法檢出2份大腸桿菌O157∶H7、14份沙門菌和21份產單核細胞李氏桿菌。食品檢測中用傳統培養法鑒定的陽性樣本與qPCR方法檢測結果一致,應用qPCR方法檢測食源性致病菌的陽性檢出率較傳統培養法略高,而多重qPCR方法的檢測時間更短。

表4 多重qPCR與國標法檢測對比試驗
3 討 論
傳統的細菌鑒定需要增殖培養、選擇性培養和生化鑒定3個步驟,是檢測食源性致病菌的“金標準”,缺點是耗時長,通常需要5~6 d,且檢測靈敏度低、無法進行定量分析[14]。目前,已報道多種食源性致病菌快速檢測方法[16],除平板檢測法外,常規檢測方法還有化學分析法和免疫分析法等,這些方法步驟繁瑣、耗時長、成本高,需要高專業水平人員操作,因此開發以分子生物學、免疫分析、生物傳感器、代謝學、核酸適配體為基礎,具有操作簡單、靈敏、快速和低成本等優點,具備檢測復雜食品樣品中致病菌的方法是主要研究方向。隨著分子生物學的發展和應用,實時熒光定量聚合酶鏈反應(qPCR)已成為一種強大的研究和診斷工具,簡便、快速、靈敏和特異是qPCR滿足核酸檢測所需的4個關鍵方面[17]。本研究參考多篇文獻報道的3種致病菌保守基因[11,14,18],以大腸桿菌O157∶H7rfbE基因、沙門菌invA基因和產單核細胞李氏桿菌hlyA基因設計3種致病菌特異性引物和探針,建立多重qPCR方法,進行特異性、靈敏性、重復性和臨床樣品檢測試驗。結果表明,建立的多重qPCR方法對3種致病菌均特異性擴增;靈敏性高,均達到104copies·μL-1,目前國內多數多重qPCR方法的研究中[14,19-20]靈敏性檢測限為102~105copies·μL-1,本研究建立的方法靈敏性符合理論預期;重復性良好,批內變異系數為0.09%~2.97%,小于5%,批間變異系數為0.23%~7.82%,小于10%;臨床樣品檢測同國標法差異小,國標法檢測陽性樣品用本研究方法檢測結果均為陽性;檢測時間短,僅1 h。
本研究建立的多重qPCR方法在食品檢測結果中顯示沙門菌檢出率略高于國標法,但所有受污染樣品都被檢出,證明建立的方法具有特異性和有效性,可用于實際檢測。微生物在自然環境中主要有活菌(viable)、活的非可培養狀態(viable but non-culturable, VBNC)、具有生物活性的死菌(ghosts)以及細胞膜損傷的死菌(membrane compromised)4種存在狀態。有研究表明[21],處于VBNC狀態時細菌不能在異養平板上生長,但仍具有新陳代謝、致病性和毒性,從而逃避國標法的檢測,成為影響食品安全的隱性污染源,qPCR法作為分子生物學方法根據致病菌的特異性基因來檢測,不區分活菌和死菌,多重qPCR法的陽性檢測率略高于國標法,多篇文獻報道[14-15,22]也證實了這一點。多重qPCR反應在同一體系下進行,大大節省了試劑和材料,檢測程序更加精準、快速[22]。綜上所述,研究建立的多重qPCR方法可作為食品中大腸桿菌O157∶H7、沙門菌和產單核細胞李氏桿菌3種食源性致病菌一種準確、快速、穩定的檢測手段。
應用本研究建立的多重qPCR方法對保定市市售生鮮肉食品中3種致病菌污染進行檢測分析,采集的120份生鮮肉樣品中,受污染樣品34份,總檢出率為28.33%,檢測出2份大腸桿菌O157∶H7、14份沙門菌和21份產單核細胞李氏桿菌,陽性檢出率分別為1.67%、11.67%和17.50%,其中有3份樣品為沙門菌和產單核細胞李氏桿菌混合感染。2010—2016年,河北省共報告食源性疾病暴發事件308起,發病3 152人,死亡25人,病死率為0.79%,其中微生物性是主要致病因素,占24.7%[23]。近年來,河北省對食源性致病菌污染研究較少,關于石家莊、滄州和邯鄲等部分地區流行情況的文獻報道顯示食源性致病菌存在不同程度污染[24-26],2015年,石家莊市市售食品食源性致病菌總檢出率為25.00%,沙門菌和產單核細胞李氏桿菌檢出率分別為4.08%和3.06%;2016—2017年,滄州市食品食源性致病菌總檢出率為22.38%;2014 年邯鄲市食品致病菌監測總檢出率為14.4%,產單核細胞李氏桿菌為6.0%,沙門菌為1.3%。綜上所述,不同地區存在不同程度的食源性致病菌污染,對食品安全造成潛在威脅,必須加強各地食源性致病菌污染監測和食品衛生管理,保障食品安全。
4 結 論
建立了能快速、準確檢測食品中大腸桿菌O157∶H7、沙門菌和產單核細胞李氏桿菌3種食源性致病菌的多重qPCR檢測方法。應用該方法對120份保定市售生鮮肉樣品進行檢測,總檢出率為28.33%,大腸桿菌O157∶H7、沙門菌和產單核細胞李氏桿菌陽性檢出率分別為1.67%、11.67%和17.50%。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
