參附注射液質(zhì)量標(biāo)準(zhǔn)研究*
2022-04-09 05:14:06袁海英史琳莉蔡幫軍侯新蓮
中國(guó)中醫(yī)急癥 2022年3期
袁海英 史琳莉 蔡幫軍 侯新蓮
[華潤(rùn)三九(雅安)藥業(yè)有限公司,四川 雅安 625000]
參附注射液是由紅參和附子2味中藥經(jīng)現(xiàn)代工藝制備而成的注射劑,比傳統(tǒng)的參附湯起效快、療效好,在臨床上廣泛用于心衰、急性心梗、休克等心血管危重疾病的搶救,其主要有效成分為人參皂苷和烏頭類生物堿[1-3]。參附注射液在制備過(guò)程中去除了多糖、蛋白質(zhì)、黏液質(zhì)、鞣質(zhì)等大分子物質(zhì),并使毒性較大的雙酯型生物堿轉(zhuǎn)化為單酯型生物堿。而參附注射液現(xiàn)行版質(zhì)量標(biāo)準(zhǔn)已有人參皂苷含量測(cè)定項(xiàng),同時(shí)對(duì)毒性較大的雙酯型生物堿含量也進(jìn)行了限度要求,為了進(jìn)一步控制參附注射液的安全性及有效性,本試驗(yàn)建立了高效凝膠滲透色譜法[4-8]對(duì)參附注射液中大分子物質(zhì)進(jìn)行定性鑒別,同時(shí)建立了高效液相色譜法[9-12]測(cè)定其有效成分單酯型生物堿的含量。
1 儀器與試藥
1.1 儀器
Agilent 1260超高效液相色譜儀(美國(guó)Agilent)、Alltech ESLD 2000型蒸發(fā)光散射檢測(cè)器、XPE26電子天平(梅特勒)、UPT-1-40L超純水器(優(yōu)普)。
1.2 試藥
參附注射液為華潤(rùn)三九(雅安)藥業(yè)有限公司生產(chǎn),批號(hào):180101010、180403010、180404010、180501010、180601010、180701010、180801010、180901010、181001010、181101010(國(guó)藥準(zhǔn)字Z51020664)。右旋糖酐系列標(biāo)準(zhǔn) 品(D0-180,D1-2500,D2-4600,D3-7100,D4-10000,D5-21400,D6-41100,D7-84400,D8-133800,D2000-2000000;批號(hào)為140637-2000-01)購(gòu)于中國(guó)食品藥品檢定研究院。苯甲酰新烏頭原堿對(duì)照品(批號(hào):111795-201303)、苯甲酰烏頭原堿對(duì)照品(批號(hào):111794-201303)、苯甲酰次烏頭原堿對(duì)照品(批號(hào):111796-201303),均購(gòu)于中國(guó)藥品生物制品檢定所。甲醇為色譜純(默克),水為超純水,其他試劑均為分析純。
2 方法與結(jié)果
2.1 大分子物質(zhì)鑒別
2.1.1 色譜條件 采用Agilent 1260型高效液相色譜儀,凝膠色譜柱為TSK gel G4000PWXL(300 mm×7.8 mm,10 μm),流動(dòng)相為50 mmol乙酸銨溶液,流速0.5 mL/min,柱溫40℃,蒸發(fā)光檢測(cè)器,漂移管溫度115℃,干燥氣流速3.2 L/min,供試品溶液進(jìn)樣量為10 μL。
2.1.2 溶液的制備 對(duì)照品溶液:取右旋糖酐標(biāo)準(zhǔn)品適量,精密稱定,分別加水制成每1毫升約含2 mg的溶液,用0.45 μm微孔濾膜濾過(guò)即得。供試品溶液:取參附注射液成品,用0.45 μm微孔濾膜濾過(guò)即得;另取參附注射液中間體,按參附注射液處方用水稀釋至配制量,混勻用0.45 μm微孔濾膜濾過(guò)即得。
2.1.3 標(biāo)準(zhǔn)曲線 分別精密稱取系列右旋糖苷標(biāo)準(zhǔn)品D1(Mp2700)、D2(Mp5250)、D3(Mp9750)、D4(Mp13050)、D5(Mp36800)、D6(Mp64650)、D7(Mp135350)、D8(Mp300600)8種對(duì)照品適量,分別加超純水溶解成2.0 mg/mL的對(duì)照品溶液。分別精密吸取各對(duì)照品溶液10 μL注入高效液相色譜儀,依法測(cè)定。以對(duì)照品相對(duì)分子質(zhì)量的對(duì)數(shù)值為橫坐標(biāo)(X),保留時(shí)間為縱坐標(biāo)(Y),繪制標(biāo)準(zhǔn)曲線。結(jié)果回歸方程為Y=-3.003X+30.415,r=0.997。表明相對(duì)分子質(zhì)量在2 700~300 600內(nèi)其對(duì)數(shù)值與保留時(shí)間呈現(xiàn)良好的線性關(guān)系。
2.1.4 檢出限測(cè)定 精密量取右旋糖酐D4標(biāo)準(zhǔn)品適量,加超純水配成2.168 mg/mL的對(duì)照品溶液,再逐級(jí)稀釋,分別取10 μL依法測(cè)定,記錄色譜圖。以信噪比約為3∶1時(shí)相應(yīng)的進(jìn)樣量0.678 μg確定為檢出限。
2.1.5 精密度試驗(yàn) 精密吸取“2.1.4”項(xiàng)下右旋糖酐D4對(duì)照品溶液,依法連續(xù)測(cè)定6次,計(jì)算6次保留時(shí)間的RSD為0.12%,峰面積對(duì)數(shù)值的RSD為0.31%,表明在儀器精密度良好。
2.1.6 穩(wěn)定性試驗(yàn) 取“2.1.4”項(xiàng)下右旋糖酐D4對(duì)照品溶液,分別于0、2、4、8、12、24 h重復(fù)測(cè)定,計(jì)算24 h內(nèi)保留時(shí)間的RSD為0.47%,峰面積對(duì)數(shù)值的RSD為0.36%,表明待測(cè)成分在24 h內(nèi)穩(wěn)定。
2.1.7 重復(fù)性試驗(yàn) 取同一批參附注射液,分別按“2.1.2”項(xiàng)下方法制備成6份供試品溶液,依法測(cè)定,均未檢出保留時(shí)間大于右旋糖酐D4的色譜峰,且樣品中其他色譜峰保留時(shí)間的RSD均小于2.0%,峰面積對(duì)數(shù)值的RSD為0.36%,說(shuō)明重復(fù)性良好。
2.1.8 樣品測(cè)定 取3批參附注射液中間體和對(duì)應(yīng)批次成品,另隨機(jī)取7批參附注射液成品,按照“2.1.2”項(xiàng)下方法制備成供試品溶液,依法測(cè)定,結(jié)果在3批中間體樣品中檢出了保留時(shí)間大于D4和D8的色譜峰,而在參附注射液成品中,則均未檢出保留時(shí)間大于D4的色譜峰,見(jiàn)圖1。說(shuō)明在生產(chǎn)工藝過(guò)程中,大分子物質(zhì)可被去除。
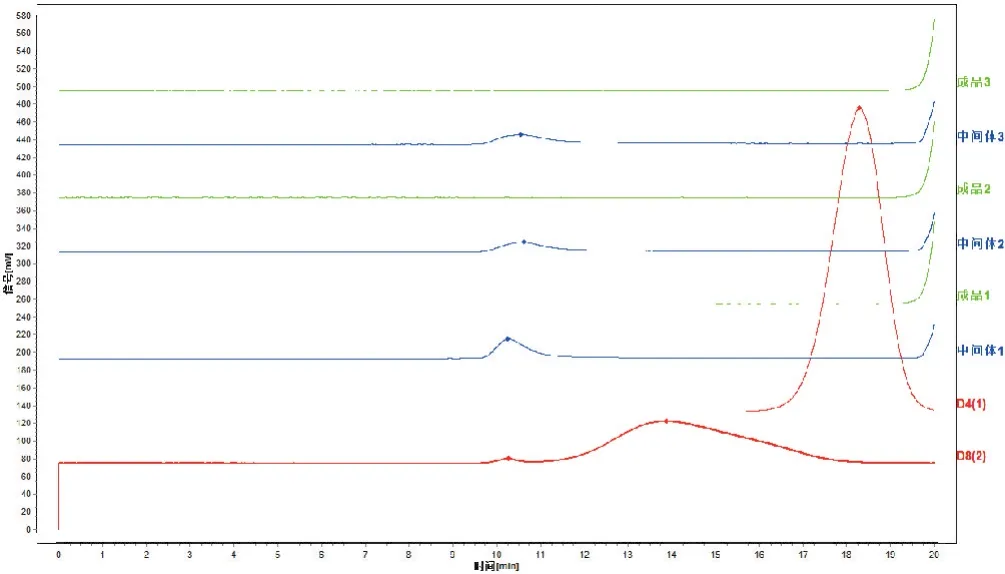
圖1 參附注射液中間體及成品大分子物質(zhì)檢測(cè)色譜圖
2.2 單酯型生物堿含量測(cè)定
2.2.1 色譜條件 采用Agilent 1260高效液相色譜儀,色譜柱 Thermo Hypersil BDS C18(4.6 mm×250 mm,5 μm),以0.2%三乙胺(用冰醋酸調(diào)至pH5.3)為流動(dòng)相A,甲醇為流動(dòng)相B,按表1中的規(guī)定進(jìn)行梯度洗脫,檢測(cè)波長(zhǎng)為235 nm;柱溫30℃;流速1.0 mL/min,進(jìn)樣量20 μL。理論塔板數(shù)按苯甲酰新烏頭原堿峰計(jì)算應(yīng)不低于20 000。

表1 梯度洗脫表
2.2.2 溶液的制備 對(duì)照品溶液:取苯甲酰新烏頭原堿對(duì)照品、苯甲酰烏頭原堿對(duì)照品、苯甲酰次烏頭原堿對(duì)照品適量,精密稱定,加異丙醇-二氯甲烷(1∶1)混合溶液分別制成每1毫升含苯甲酰新烏頭原堿509.62 μg、苯甲酰烏頭原堿507.82 μg、苯甲酰次烏頭原堿503.88 μg的對(duì)照品貯備溶液。再精密吸取上述各對(duì)照貯備溶液1 mL至50 mL量瓶中,加異丙醇-二氯甲烷(1∶1)的混合溶液至刻度,搖勻制成每1毫升含苯甲酰新烏頭原堿、苯甲酰烏頭原堿、苯甲酰次烏頭原堿分別為10.19、10.16、10.08 μg的混合對(duì)照品溶液。供試品溶液制備:取參附注射液10 mL,加于固相萃取小柱(Phenomenex Strata-X,先以甲醇15 mL活化,再以15 mL水平衡),緩慢過(guò)柱,以水5 mL沖洗,再以甲醇緩慢洗脫,用1 mL量瓶收集甲醇洗脫液至刻度,搖勻作為供試品溶液。另按參附注射液工藝制備缺附子的陰性樣品,照供試品處理方法制備成陰性樣品溶液。
2.2.3 專屬性試驗(yàn) 精密吸取混合對(duì)照品溶液、供試品溶液和陰性樣品溶液各20 μL,按“2.2.1”項(xiàng)下色譜條件測(cè)定,結(jié)果陰性樣品對(duì)供試品測(cè)定無(wú)干擾,見(jiàn)圖2,說(shuō)明本方法專屬性良好。
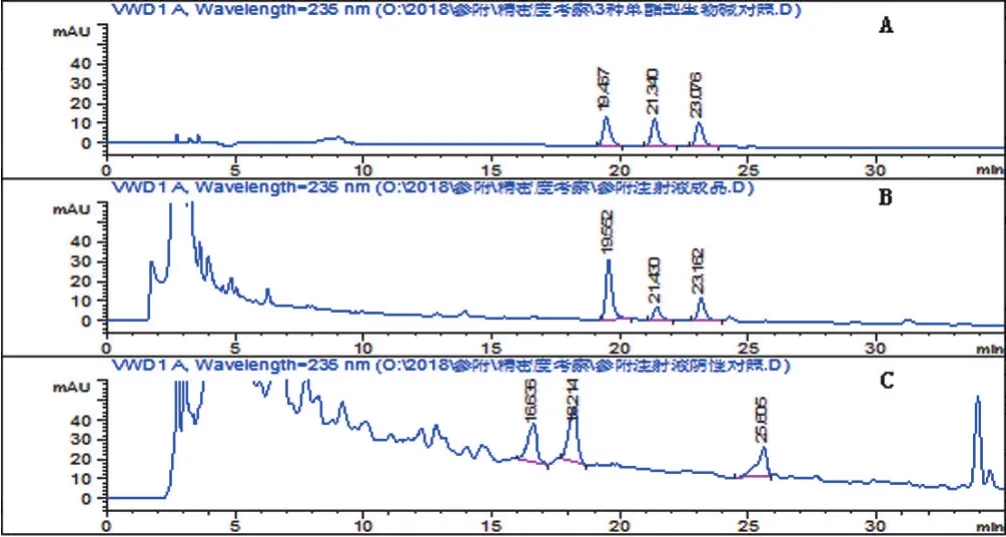
圖2 對(duì)照品(A)、供試品(B)和陰性樣品(C)HPLC色譜圖
2.2.4 精密度試驗(yàn) 精密吸取“2.2.2”項(xiàng)下混合對(duì)照品溶液20 μL,連續(xù)進(jìn)樣6次,測(cè)得苯甲酰新烏頭原堿峰面積RSD為1.78%、苯甲酰烏頭原堿峰面積RSD為0.99%、苯甲酰次烏頭原堿峰面積RSD為1.34%,表明儀器精密度良好。
2.2.5 線性關(guān)系考察 精密量取“2.2.2”項(xiàng)下各對(duì)照品貯備溶液2 mL至同一10 mL量瓶中,用異丙醇-二氯甲烷(1∶1)的混合溶液稀釋定容至刻度,搖勻制成每1 mL含苯甲酰新烏頭原堿、苯甲酰烏頭原堿、苯甲酰次烏頭原堿分別為101.92 μg、101.56 μg、100.77 μg的混合對(duì)照溶液。再將該混合對(duì)照溶液用異丙醇-二氯甲烷(1∶1)的混合溶液逐級(jí)稀釋2倍、5倍、10倍、20倍、40倍。精密吸取各濃度混合對(duì)照品溶液20 μL,按“2.2.1”項(xiàng)下色譜條件依法檢測(cè),以進(jìn)樣量(X)為橫坐標(biāo),色譜峰峰面積為縱坐(Y,μg)標(biāo)繪制標(biāo)準(zhǔn)曲線,各成分回歸方程、線性范圍見(jiàn)表2。結(jié)果表明各成分在線性范圍內(nèi)與峰面積有良好的線性關(guān)系。

表2 線性關(guān)系考察結(jié)果
2.2.6 重復(fù)性試驗(yàn) 取同一批參附注射液,按“2.2.2”項(xiàng)下方法平行制備供試品溶液6份,按“2.2.1”項(xiàng)下色譜條件測(cè)定,結(jié)果苯甲酰新烏頭原堿平均含量為1.85 μg/mL,RSD為1.69%;苯甲酰烏頭原堿平均含量為0.61 μg/mL,RSD為1.23%;苯甲酰次烏頭原堿平均含量為1.23 μg/mL,RSD為0.98%。表明本方法具有良好的重復(fù)性。
2.2.7 穩(wěn)定性試驗(yàn) 取同一參附注射液供試品溶液,室溫放置0、2、4、8、12、24 h,按“2.2.1”項(xiàng)下色譜條件測(cè)定,結(jié)果24 h內(nèi)苯甲酰新烏頭原堿平均含量為1.86 μg/mL,RSD為1.17%;苯甲酰烏頭原堿平均含量為0.62 μg/mL,RSD為1.94%;苯甲酰次烏頭原堿平均含量為1.24 μg/mL,RSD為0.88%。表明在24 h內(nèi)供試品溶液穩(wěn)定性良好。
2.2.8 加樣回收率試驗(yàn) 根據(jù)重復(fù)性試驗(yàn)已知參附注射液中苯甲酰新烏頭原堿、苯甲酰烏頭原堿、苯甲酰次烏頭原堿的含量,加水配制成相應(yīng)濃度的混合對(duì)照品溶液,精密吸取5 mL同一批參附注射液共6份,分別精密加入相應(yīng)濃度的混合對(duì)照品溶液各5 mL,按“2.2.2”項(xiàng)下方法制備供試品溶液,再按“2.2.1”項(xiàng)下色譜條件測(cè)定,結(jié)果見(jiàn)表3。苯甲酰新烏頭原堿、苯甲酰烏頭原堿、苯甲酰次烏頭原堿的加樣回收率均在85%~110%之間,RSD均小于3%,符合《中國(guó)藥典》相關(guān)要求,表明本方法具有較好的準(zhǔn)確度。

表3 加樣回收試驗(yàn)結(jié)果
2.2.9 樣品測(cè)定 取10批參附注射液,按“2.2.2”項(xiàng)方法制備供試品溶液,并按“2.2.1”項(xiàng)下色譜條件測(cè)定,測(cè)定結(jié)果見(jiàn)表4。
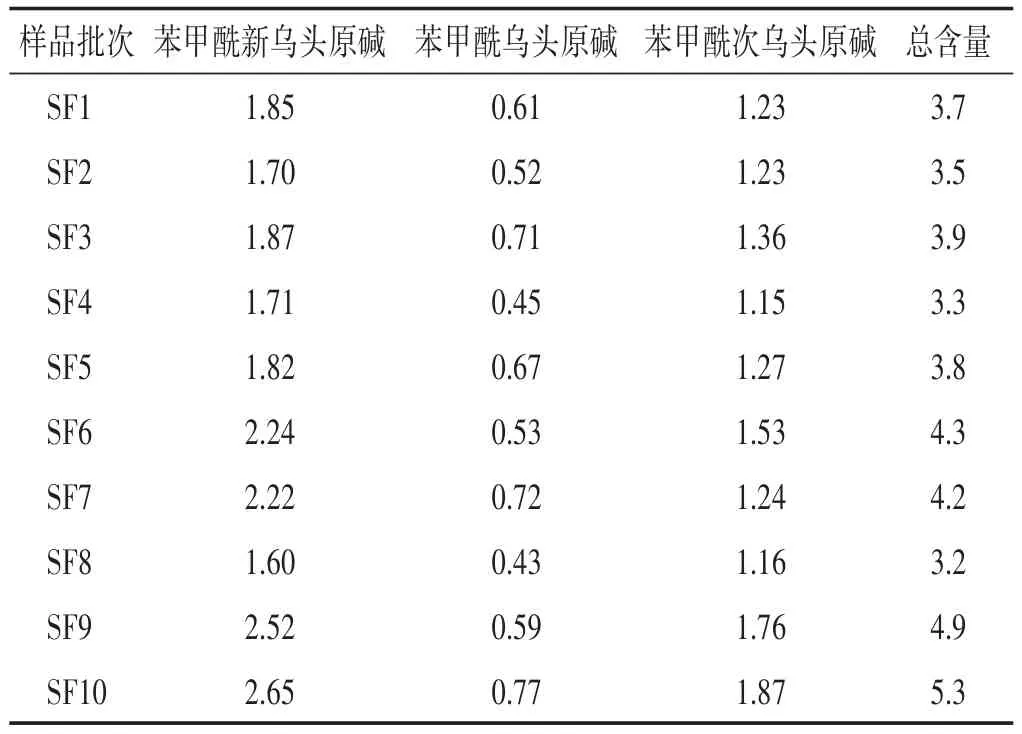
表4 參附注射液3種單酯型生物堿含量測(cè)定結(jié)果(μg/mL)
3 結(jié) 論
中藥注射劑經(jīng)過(guò)幾十年的發(fā)展,在提升藥效的同時(shí)不良反應(yīng)也時(shí)有發(fā)生,其安全性得到廣泛關(guān)注。據(jù)文獻(xiàn)報(bào)道,影響中藥注射劑安全性的主要物質(zhì)基礎(chǔ)是大分子物質(zhì)[13-16],包括蛋白質(zhì)、鞣質(zhì)、多糖、樹(shù)脂等,本研究建立了參附注射液中大分子物質(zhì)定性鑒別方法,有利于監(jiān)控生產(chǎn)過(guò)程中大分子物質(zhì)的去除情況,對(duì)控制產(chǎn)品質(zhì)量具有重要意義,對(duì)其他中藥注射劑中大分子物質(zhì)的鑒別也有參考作用。故在參附注射液原注冊(cè)標(biāo)準(zhǔn)的基礎(chǔ)上增加大分子物質(zhì)檢查項(xiàng),規(guī)定在色譜圖中不得檢出保留時(shí)間小于標(biāo)準(zhǔn)品右旋糖酐D4保留時(shí)間的色譜峰。
本研究建立了參附注射液處方中另一味藥材附子中有效成分單酯型生物堿的含量測(cè)定方法。通過(guò)對(duì)10批參附注射液中3種單酯型生物堿總含量進(jìn)行測(cè)定,結(jié)果3種單酯型生物堿總含量在3.2~5.3 μg/mL范圍內(nèi)波動(dòng),均值為4.0 μg/mL,RSD為17.0%。故增加了參附注射液中3種單酯型生物堿總含量標(biāo)準(zhǔn)為2.0~6.0 μg/mL,以此作為現(xiàn)行質(zhì)量標(biāo)準(zhǔn)的補(bǔ)充。進(jìn)一步控制參附注射液質(zhì)量穩(wěn)定性及臨床用藥的安全有效性。另在原標(biāo)準(zhǔn)中對(duì)于3種雙酯型生物堿即新烏頭堿、烏頭堿、次烏頭堿已采用薄層鑒別的方法進(jìn)行了限量檢查,需繼續(xù)進(jìn)行研究后將該方法進(jìn)行改進(jìn)。
