注射用克林霉素磷酸酯質量分析
2022-03-18 06:10:42李香荷張冬王強王韻田蘭高燕霞
中國抗生素雜志 2022年2期
李香荷 張冬 王強 王韻 田蘭 高燕霞
(河北省藥品醫療器械檢驗研究院,石家莊 050200)
克林霉素磷酸酯(clindamycin phosphate)為化學半合成的克林霉素前藥,在體外無抗菌活性,進入機體后在堿性磷酸酯酶作用下水解為克林霉素而發揮抗菌活性[1]。克林霉素作用于敏感菌核糖體的50 S亞基,抑制細菌細胞的蛋白質合成,可作為對β-內酰胺類抗生素過敏時的替代藥物[2]。臨床主要用于治療革蘭陽性菌所致的呼吸道、耳鼻喉系統、皮膚軟組織、泌尿系統等感染,還可用于厭氧菌引起的各種感染性疾病。不良反應主要為腎功能損害和血尿。
克林霉素磷酸酯注射液為美國法瑪西亞普強公司(Pharmacia and Upjohn Company,即現在的輝瑞制藥)研發,在1982年上市,其規格為2 mL:0.3 g。1994年,華北制藥廠的仿制藥被批準在國內上市。目前克林霉素磷酸酯制劑國產批準文號258個,劑型包括:膠囊劑、片劑、注射劑、凝膠劑、栓劑和陰道乳膏劑。注射用克林霉素磷酸酯批準文號103個,生產企業36家,規格有:0.15、0.25、0.3、0.4、0.45、0.5、0.6、0.75、0.9和1.2 g。
克林霉素磷酸酯原料藥收載于《中國藥典》2020版、USP44、EP11.0、BP2021和JP2021,注射用粉針劑各國藥典均未收載。目前國內執行國家藥品標準有:WS1-(X-358)-2003Z-2011-2017、WS1-(X-526)-2003Z-2010、YBH02202016、YBH00802012、YBH03602014、YBH02292015等。2021年,該品種被列為國家藥品評價性抽驗品種。共抽取142批次,涉及21個生產企業和38個批準文號,見表1。本文參考文獻報道[3-9],通過對國產注射用克林霉素磷酸酯的法定標準檢驗及探索性研究,以問題為導向,以提高藥品質量為目標,對該品種藥品進行全面質量評價。
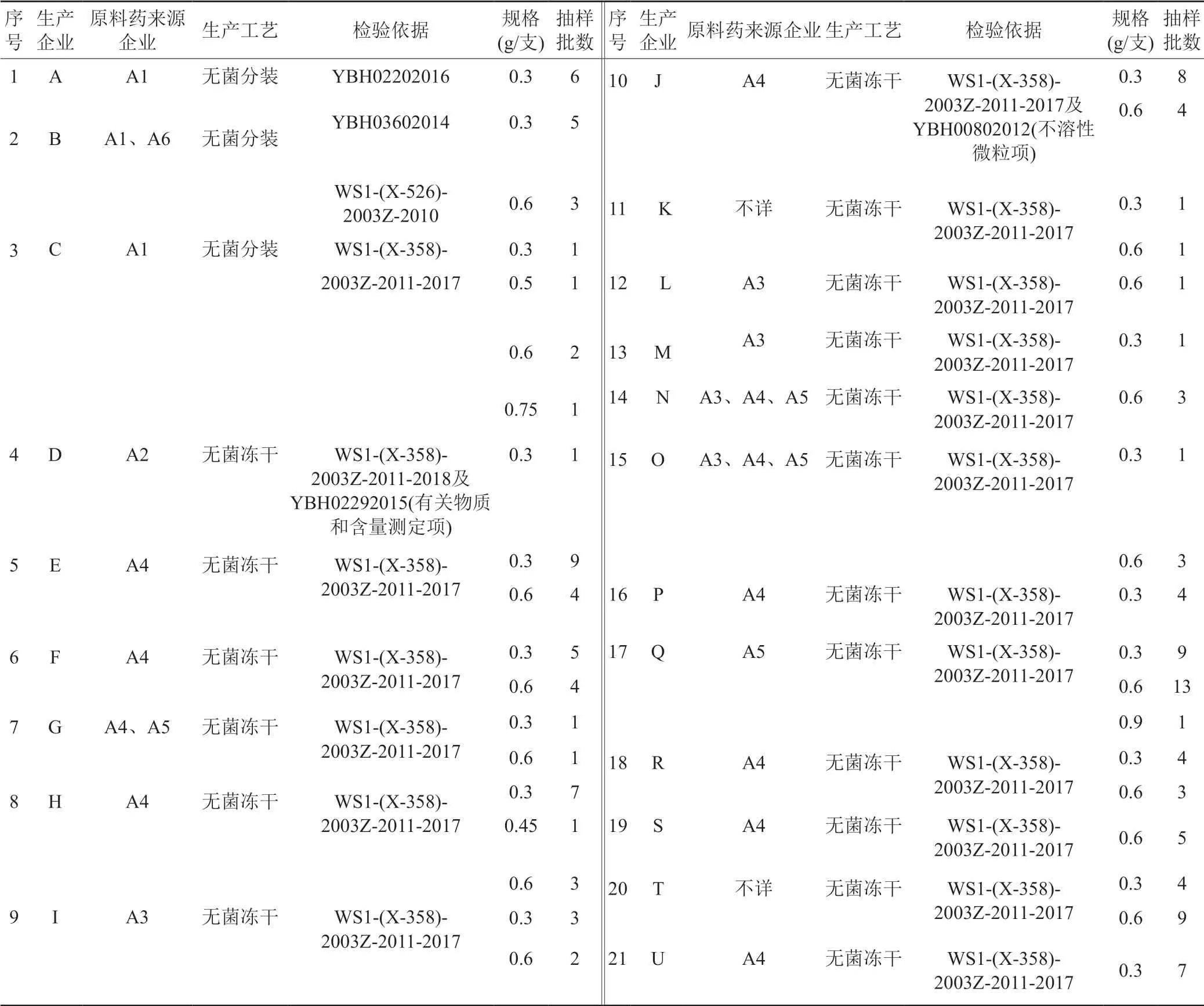
表1 抽樣情況表Tab.1 Table of samping
1 材料與方法
1.1 儀器
島津離子阱飛行時間液質聯用儀,安捷倫1260高效液相色譜儀;梅特勒XS125分析天平,XSPBM-3CA 顯微鏡,布魯克D2 Phaser X射線衍射儀。
1.2 試藥
克林霉素磷酸酯對照品(中檢院提供,批號130486-201805,含量82.7%);醋酸銨、甲醇、乙腈均為色譜純(Fisher chemical),其余試劑為分析純。
142批次注射用克林霉素磷酸酯為2021年國家評價性抽驗樣品,規格分別為0.3、0.45、0.5、0.6、0.75和0.9 g/支。其中3個企業采用無菌分裝工藝,共抽樣19批,18個企業采用無菌凍干工藝,共抽樣123批,調研收集到19個企業的原料和輔料,其對應的原料涉及6家企業。
2 試驗方法
2.1 法定檢驗
按產品說明書上對應的執行標準,對142批次注射用克林霉素磷酸酯進行法定標準檢驗,主要評價項目為有關物質、水分、溶液的澄清度與顏色、酸度和含量測定。
2.2 HPLC法分析有關物質
色譜條件:色譜柱為島津Shim-pack C18柱(規格:4.6 mm×250 mm,5 μm);流動相A:pH6.0磷酸鹽緩沖液(稱取磷酸二氫鉀13.6 g,加水1000 mL使溶解,用0.45 mg/mL氫氧化鉀溶液調節pH值至6.0)-乙腈(79:21,V/V),流動相B:pH6.0磷酸鹽緩沖液(稱取磷酸二氫鉀13.6 g,加水1000 mL使溶解,用0.45 mg/mL氫氧化鉀溶液調節pH值至6.0)-乙腈(40:60,V/V),先以流動相A等度洗脫,待克林霉素磷酸酯峰洗脫完畢后立即按表2線性梯度洗脫;流速:1.1 mL/min;柱溫:35℃;進樣量:20 μL;檢測波長:210 nm。
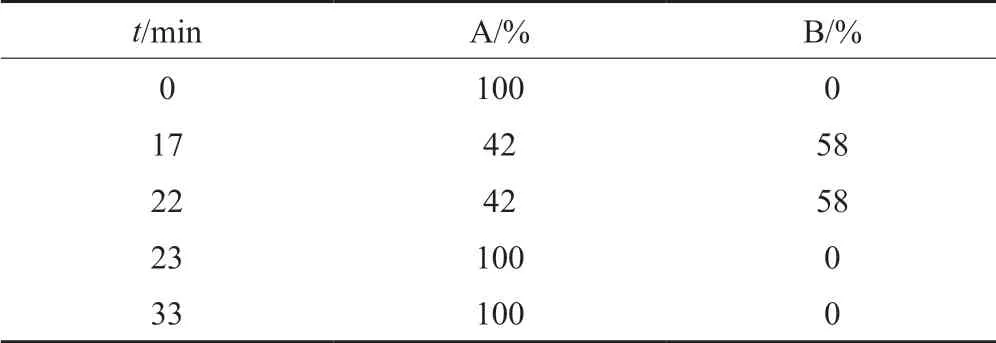
表2 梯度洗脫程序Tab.2 Gradient elution program
2.3 LC-MS分析雜質結構
液相色譜條件:色譜柱為島津Shim-pack C18柱(規格:4.6 mm×250 mm,5 μm);流動相A:0.036 mol/L乙酸銨溶液(用乙酸調節pH值至6.0),流動相B:乙腈;按表3線性梯度洗脫;柱溫35 ℃;流速:1.0 mL/min(柱后分流比為7:3,V/V);進樣體積20 μL。
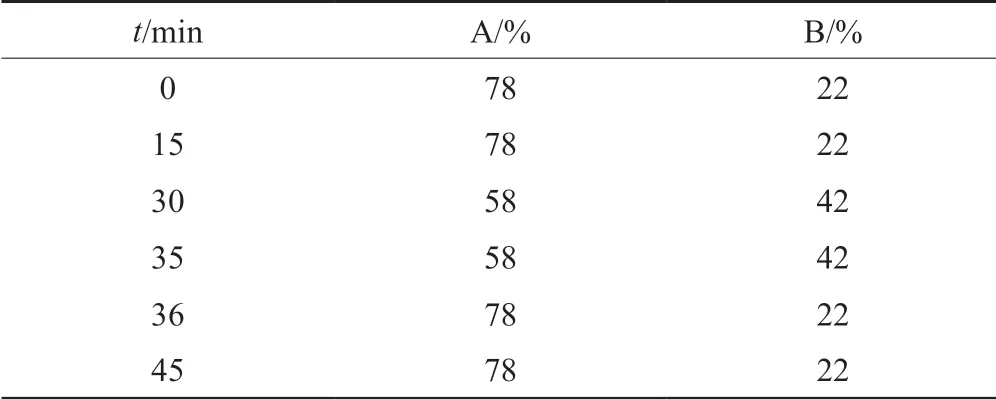
表3 梯度洗脫程序Tab.3 Gradient elution program
質譜條件:離子源:ESI(+);檢測電壓:1.54 kV;CDL溫度:200℃;脫溶劑管溫度:200℃。
2.4 HPLC含量測定
色譜條件:色譜柱為島津Shim-pack C18柱(規格:4.6 mm×250 mm,5 μm);流動相:pH6.0磷酸鹽緩沖液(稱取磷酸二氫鉀13.6 g,加水1000 mL使溶解,用0.45 mg/mL氫氧化鉀溶液調節pH值至6.0)-乙腈(79:21,V/V);柱溫:35℃;流速:1.1 mL/min;進樣體積20 μL;檢測波長為210 nm。
3 結果與討論
3.1 法定標準檢驗
依據法定標準檢驗,142批次抽樣,全部符合規定,合格率為100%。對與產品質量密切相關的主要檢驗項目如有關物質、水分、溶液的澄清度與顏色、酸度和含量測定結果進行統計分析。
3.1.1 有關物質
現行法定標準中的有關物質檢測方法及限度差異較大。比較其差異:WS1-(X-358)-2003Z-2011-2017、YBH03602014(B企業 0.3 g)和YBH02292015(D企業)標準,采用梯度系統(色譜系統相同),YBH02202016(A企業)、WS1-(X-526)-2003Z-2010(B企業0.6 g)標準采用等度法。在限度方面,差異主要集中于對已知雜質的控制,國家藥品標準WS1-(X-358)-2003Z-2011-2017僅控制林可霉素、克林霉素兩個已知雜質;YBH02202016(A企業)控制了7個已知雜質;YBH02292015(D企業)控制了8個已知雜質;YBH03602014(B企業0.3 g)控制了4個已知雜質,國家藥品標準WS1-(X-526)-2003Z-2010(B企業0.6g)未控制已知雜質,僅控制了單雜和總雜。
139批次中均未檢出林可霉素,克林霉素含量為0~0.6%,其他單雜含量為0.01%~1.4%,其他總雜含量為0.1%~3.6%;不同工藝產品之間雜質差異較大(圖1),其中企業A、B和C 3家無菌分裝產品,克林霉素含量均值分別為0.015%、0.10%和0.08%、其他單雜含量均值分別為0.03%、0.11%和0.38%,其他總雜含量均值分別為0.11%、0.30%和0.62%;M企業(凍干工藝)產品雜質含量最高,克林霉素含量為0.6%、其他單雜含量為1.2%,其他總雜含量為3.1%。采用無菌分裝的3家企業產品無論單個雜質還是雜質總量均遠低于凍干工藝產品。
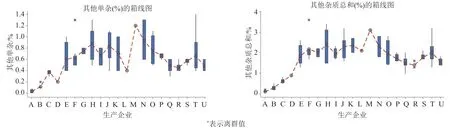
圖1 有關物質檢驗結果Fig.1 The standard test results for related substances
同一生產企業不同批次產品之間亦存在一定的差異。統計同企業3批次以上的產品結果,其他總雜含量差異最大的產品來自A、B、O、H和T企業,其中A和B的產品(無菌分裝品),由于其雜質含量本身較低,使得批間的RSD相對較大;O、H和T企業產品(無菌凍干品),由于穩定性較差,其雜質含量與貯存時間呈正相關(P<0.05),貯存時間的差異導致批間雜質含量差異較大,使得RSD值偏大。
此外,現行質控標準的差異使得測定結果可比性不強。從測定結果看,不同分析方法造成雜質總量的極差可高達3.5%,說明該品種有關物質控制方法亟待規范統一。
3.1.2 水分
各現行標準中的水分限度不統一。國家藥品標準WS1-(X-358)-2003Z-2011-2017和YBH03602014(B企業)均規定“水分不得過1.5%”,YBH02202016(A企業)規定“水分不得過6.0%”,國家藥品標準WS1-(X-526)-2003Z-2010(B企業0.6g)規定“水分不得過1.3%”。
142批次樣品采用費休氏法測定水分,結果為0.2%~1.4%,均值為0.77%,企業間水分結果差異較大(圖2),其中,A、B、C、K和R企業5家的產品水分含量均值較低,分別為0.4%、0.4%、0.5%、0.3%和0.3%;I、O、P和F企業4家的產品水分值較高,分別為1.2%、1.1%、1.2%和1.1%。3家無菌分裝工藝的產品水分含量明顯低于采用無菌凍干工藝的產品;不同企業凍干工藝產品的水分含量差異較大,其中E、O、S和T企業產品的批間差異較大,反映該企業凍干工藝穩定性有待提高。
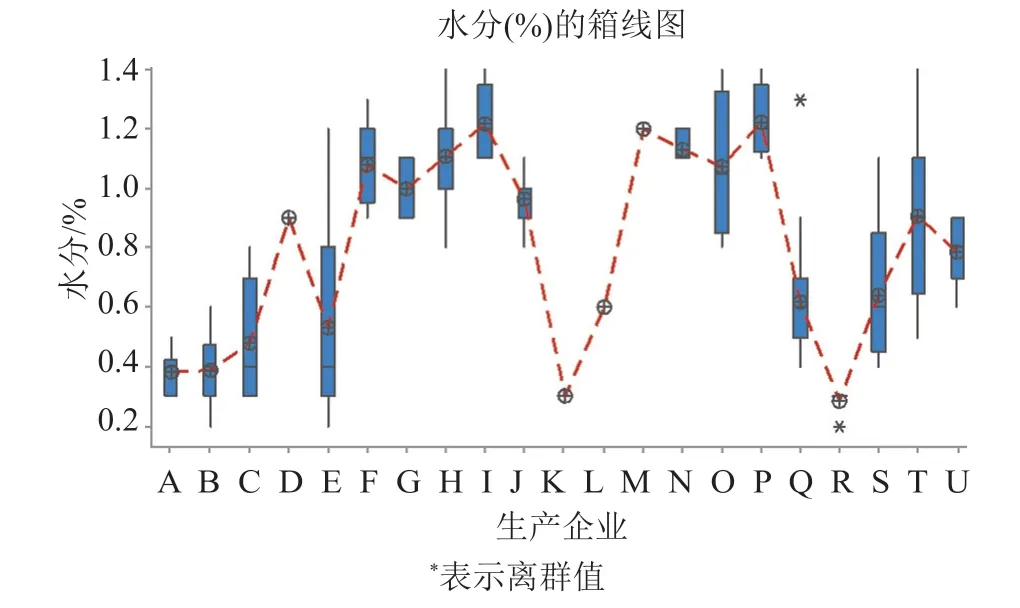
圖2 水分檢測結果Fig.2 The result of moisture
3.1.3 溶液的澄清度與顏色
凍干工藝產品的檢測濃度為0.2 g/mL,無菌分裝產品的檢測濃度為0.04 g/mL;142批次產品檢驗結果均符合規定,但檢測濃度的差異使得其結果不完全具有可比性。
檢驗結果顯示,137批次產品澄清,5批次澄清度略顯渾濁,但淺于1號濁度標準液;140批次產品的溶液顏色為無色,S企業和J企業各1批產品顏色稍深,介于黃色1號與2號之間。
3.1.4 酸度
按法定標準檢驗均符合規定。其中,123批次無菌凍干產品的pH值在6.2~6.8之間(標準規定pH值5.0~7.0),19批次無菌分裝產品的pH值在3.7~4.4之間(標準規定pH值3.5~4.5),同一生產企業不同批次的酸度值均較為接近。
3.1.5 含量測定
按法定標準檢驗142批次樣品均符合規定。雖然不同標準中含量的限度有所差異,但從所有樣品的含量測定結果,按無水物計分布于76.4%~85.8%之間,均值為80.6%,極差9.4% ;按平均裝量計,含克林霉素為標示量的94.0%~106.2%,均值為98.6%,極差12.2%;未發現低限投料的問題。
對不同生產企業3批次以上產品數據進行分析,批間含量差異最大的為B企業,變異系數為1.8%(按無水物計)和3.5%(按平均裝量計),批間含量差異最小的為O企業,變異系數為0.6%(按無水物計)和0.5%(按平均裝量計)。
3.2 探索性研究
3.2.1 雜質譜分析
采用新建立的雜質譜分析方法共檢出13個雜質,應用LC-IT-TOF/MS技術,結合克林霉素磷酸酯EP系統適用性對照品、雜質F(林可霉素2-磷酸酯)、雜質G(2,4-磷酯酰林可霉素)、雜質B(克林霉素B磷酸酯)對照品,中國藥品檢驗研究院提供的雜質A(林可霉素)和雜質E(克林霉素)對照品,參考國外藥典,確證樣品中11個雜質的結構。雜質1為林可霉素2-磷酸酯,雜質2為2,4-磷酯酰林可霉素,雜質3為林可霉素,雜質4為克林霉素2,4-二磷酸酯,雜質5為克林霉素B磷酸酯,雜質6為克林霉素2,3-二磷酸酯,雜質7為7-表克林霉素磷酸酯,雜質8為克林霉素(2R-順式)-非對映異構體2磷酸鹽,雜質9為3′(6′)-去氫克林霉素磷酸酯,雜質10為克林霉素,雜質11為克林霉素焦磷酸酯。各雜質結構見表4。
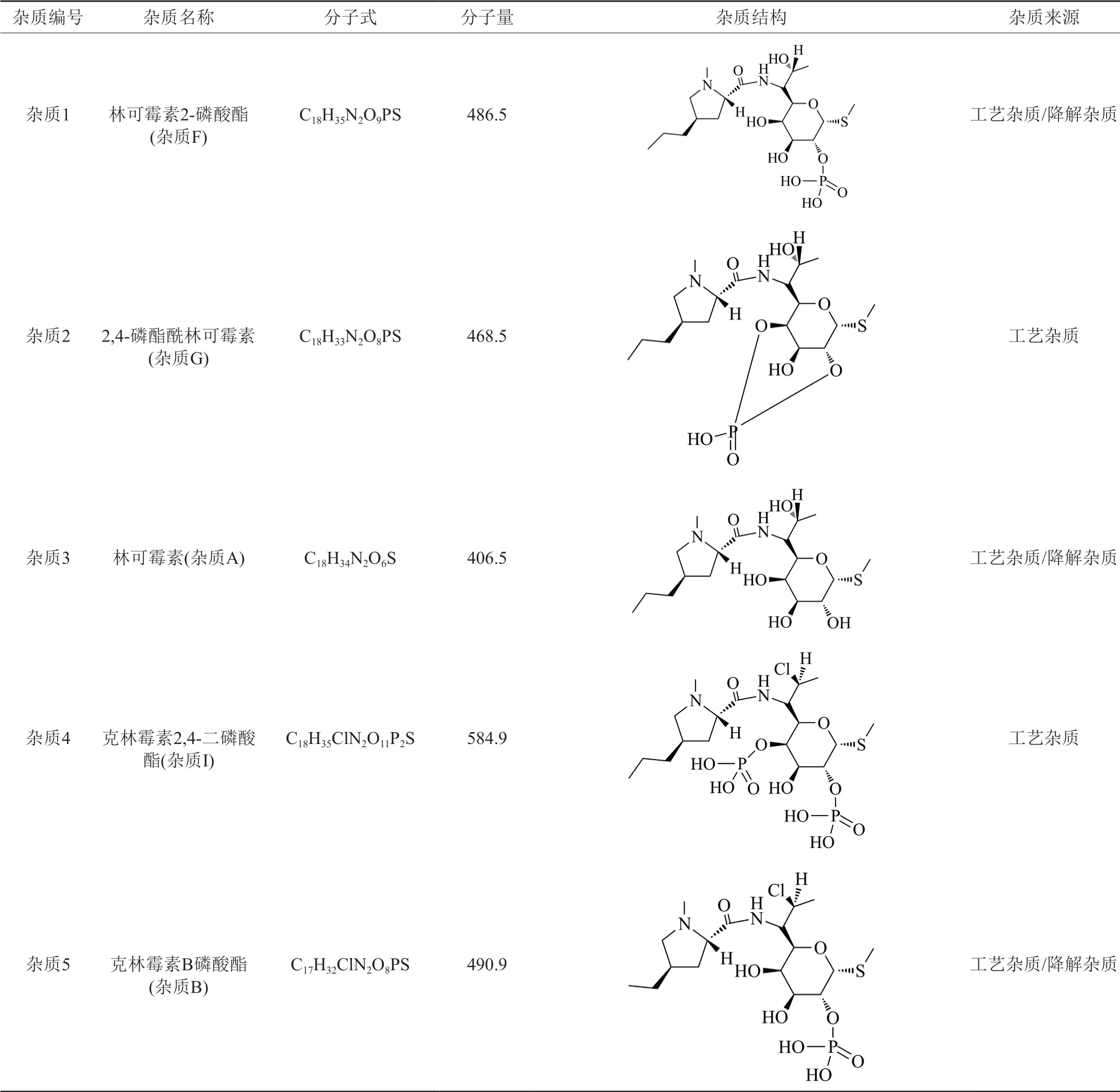
表4 各雜質推定結構及來源分析Tab.4 Structure and source analysis of peaks of related substances

續表4
利用強制降解試驗結合LC-MS/ MS質譜分析,探討產品中諸雜質的來源。無菌凍干制劑中雜質F和雜質B的含量遠高于原料,尤其是隨著貯存時間的延長,兩個雜質含量顯著增加,提示其為貯存中的主要降解雜質;克林霉素磷酸酯對酸較穩定,在熱、堿、氧化和紫外條件下易產生雜質F、G、E。
3.2.2 有關物質測定結果
比較EP11.0(BP2021與EP11.0方法及限度一致)、USP44和《中國藥典》2020年版克林霉素磷酸酯原料的有關物質方法,初步認為EP11.0收載的克林霉素磷酸酯有關物質的分析方法較理想,以此為基礎對其梯度洗脫程序進行了優化(圖3),全部已知雜質均可以良好分離。
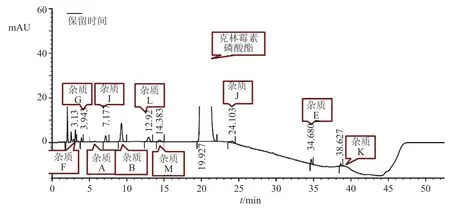
圖3 優化的有關物質高效液相色譜圖Fig.3 Optimized chromatogram of the related substances
按優化方法檢驗,142批次注射用粉針劑中,雜質F在0.04%~1.5%之間,雜質G在0.03%~0.4%之間,雜質B在0.1%~1.8%之間,雜質L在0.02%~0.6%之間,雜質E在0.03%~0.8%之間,其他單一雜質在0.1%~0.3%之間,雜質總和在0.6%~5.5%之間。
參照EP11.0、克林霉素磷酸酯原研企業的限度,建議注射用克林霉素磷酸酯有關物質限度為:雜質F(相對保留時間為0.15)不得過3.0%、雜質G(相對保留時間為0.20)不得過0.5%,雜質B(相對保留時間0.45為)不得過1.5%,雜質L(相對保留時間為0.63)不得過0.8%、雜質E(相對保留時間為1.7)不得過1.5%,其他單一雜質不得過0.5%,雜質總和不得過6.0%。142批次注射用克林霉素磷酸酯中,僅3批次近效期的冷凍干燥產品(分別來自H、T和F企業)達不到上述要求(圖4)。
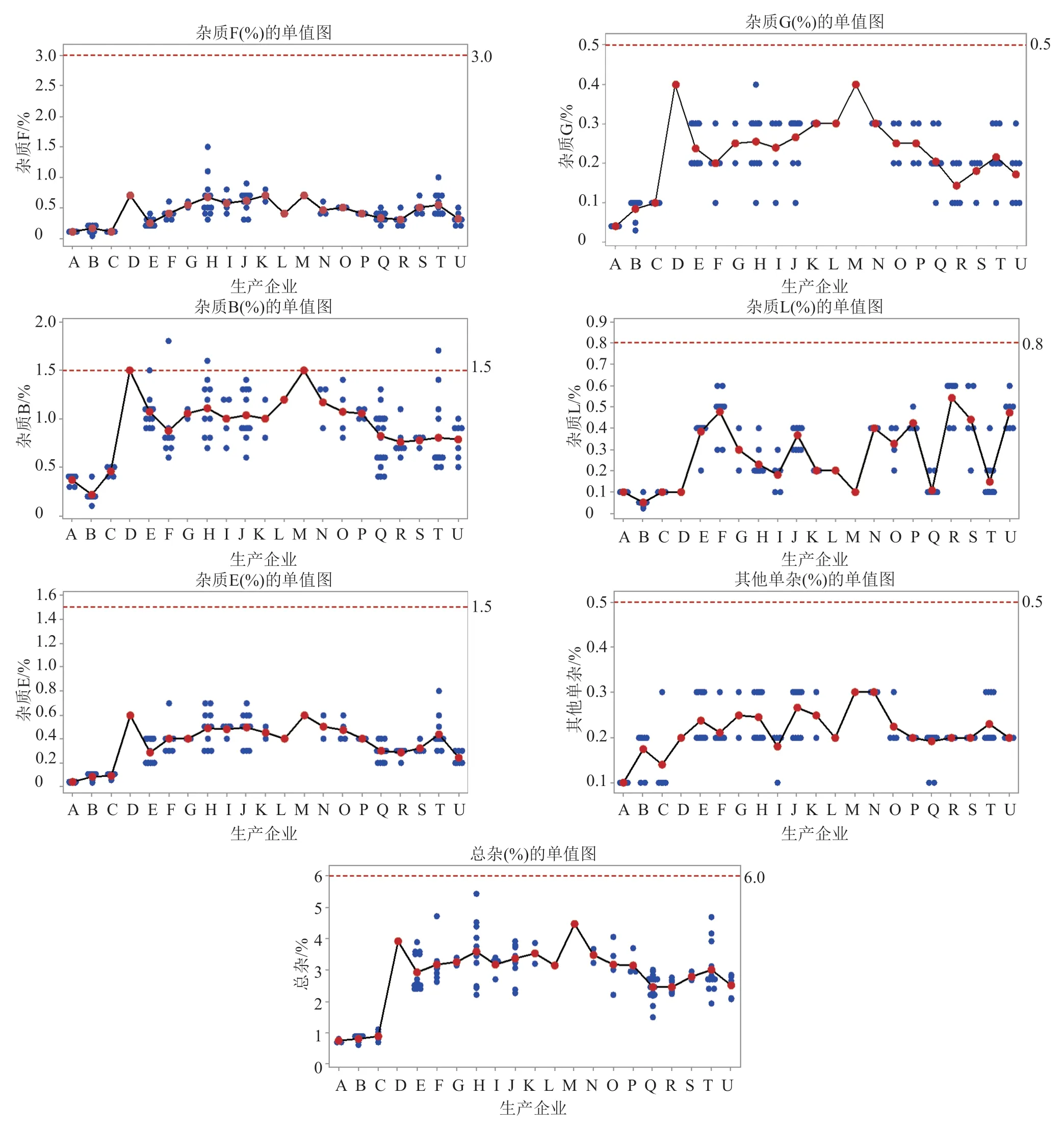
圖4 有關物質檢測結果單值圖Fig.4 The results of related substances
3.2.3 含量測定方法的改進
法定標準中的含量測定方法均采用C8色譜系統,但流動相系統與有關物質測定的流動相完全不同,結果雜質A(相對保留時間為0.97)與克林霉素磷酸酯主峰之間與不能達到完全分離(圖5),采用上述優化的有關物質分析方法測定克林霉素磷酸酯的含量,諸雜質及輔料和溶劑均不干擾測定。142批次樣品按優化后色譜系統檢驗,結果與法定標準檢驗結果相比,差異較小(圖6),但結果的穩定性更好。
圖5 法定標準典型色譜圖Fig.5 The typical chromatogram with legal standard
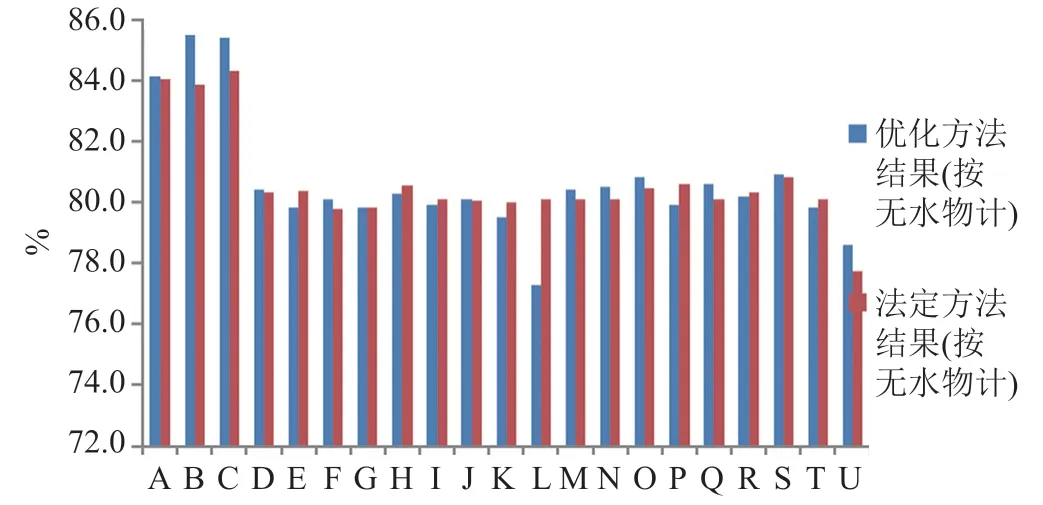
圖6 含量測定結果比較Fig.6 Comparison chart of assay results
3.2.4 原料晶型比較
目前國內克林霉素磷酸酯原料藥生產廠家為10個,調研共收集到其中6個企業(A1~A6企業)的原料。采用顯微鏡觀察法對6個企業原料晶型進行測定,結果 A1企業的晶粒規整,有六邊形板狀和不規整狀,細晶少,A2、A3和A4企業的晶粒為長方體板狀,A5和A6企業的晶粒較小,且晶體粒徑大小不一(圖7)。

圖7 克林霉素磷酸酯原料晶型圖Fig.7 The pictures of clindamycin phosphate
采用X-粉末衍射法對6家企業的原料進行測定,發現A6企業的原料X-粉末衍射圖峰形和其他5家企業有一定差異。提示不同原料藥企業的結晶工藝存在差異。3家采用無菌分裝工藝生產的企業所用原料均來自A1和A6企業;采用凍干工藝的產品原料來自其他4家原料企業,有的制劑企業未固定原料供應商(使用2~3家供應商的原料)。由于結晶工藝可直接影響原料的雜質譜,采用不同供應商的原料藥生產制劑,可能是導致制劑雜質譜變異的原因之一。
評價結果顯示,采用無菌分裝工藝生產的制劑,從雜質控制角度明顯優于冷凍干燥工藝制劑。但另一方面,從注射劑無菌保障水平角度,冷凍干燥工藝較無菌分裝工藝在制劑過程更易控制產品的污染。對該類產品生產工藝的評價將是國內仿制藥一致性評價/再評價的關鍵。
猜你喜歡
當代水產(2022年8期)2022-09-20 06:44:30
現代裝飾(2022年4期)2022-08-31 01:39:32
現代裝飾(2022年3期)2022-07-05 05:55:06
當代水產(2022年6期)2022-06-29 01:11:44
當代水產(2022年5期)2022-06-05 07:55:06
當代水產(2022年3期)2022-04-26 14:27:04
當代水產(2022年2期)2022-04-26 14:25:10
云南畫報(2020年9期)2020-10-27 02:03:26
Coco薇(2015年1期)2015-08-13 02:23:50
玩具(2009年10期)2009-11-04 02:33:14
