反芻動物瘤胃噬菌體的宏基因組學研究方法及進展
2022-03-08 02:34:18吳祎程周傳社譚支良
畜牧獸醫(yī)學報 2022年1期
關鍵詞:研究
吳祎程,冉 濤,周傳社*,譚支良
(1.中國科學院亞熱帶農業(yè)生態(tài)研究所 亞熱帶農業(yè)生態(tài)過程重點實驗室 畜禽養(yǎng)殖污染控制與資源化技術國家工程實驗室 湖南省動物營養(yǎng)與生理代謝實驗室 農業(yè)部中南動物營養(yǎng)與飼料科學觀測站,長沙 410125; 2. 中國科學院大學,北京 100049;3.蘭州大學草地農業(yè)科技學院,蘭州 730020)
病毒(virus)廣泛存在于地球各種環(huán)境中,是地球上分布最廣、數(shù)量最為巨大的生命實體[1]。研究發(fā)現(xiàn),大部分病毒能特異性侵染細菌,這些病毒被稱作噬菌體(bacteriophage或phage)[2-3]。在噬菌體被發(fā)現(xiàn)后不久,研究人員便證實了反芻動物瘤胃內也存在噬菌體[4-5]。長期以來,盡管研究人員意識到病毒可能在反芻動物瘤胃微生態(tài)系統(tǒng)中發(fā)揮著重要作用,但受限于技術手段和瘤胃環(huán)境的復雜性,瘤胃環(huán)境中噬菌體研究進展相對緩慢[6]。目前,對瘤胃噬菌體的認知多來自于傳統(tǒng)的培養(yǎng)、顯微鏡觀察和單一噬菌體的基因組學研究[7-8],對其在瘤胃微生態(tài)系統(tǒng)層面的認識仍十分匱乏且片面,以致至今對其在瘤胃微生態(tài)系統(tǒng)中的組成和功能仍知之甚少。近年來,隨著宏基因組學技術的發(fā)展,極大地推動了海洋生態(tài)環(huán)境中病毒生態(tài)學研究的發(fā)展,并取得了引人注目的成績[9]。宏基因組學也被逐步運用于胃腸道病毒組(virome)的研究,同時刷新了研究人員對胃腸道噬菌體的復雜性和多樣性的認知[10]。Manrique等[11]首次利用病毒宏基因組學技術發(fā)現(xiàn)腸道中81%~93%的噬菌體為新型噬菌體,由于病毒缺乏合適的標記基因和分析基準,它們無法被歸類或找到對應宿主。瘤胃微生態(tài)環(huán)境復雜,瘤胃作為一個巨大的基因資源庫,具有巨大的新病毒發(fā)掘空間,病毒宏基因組學技術與傳統(tǒng)技術的聯(lián)用在這一方面具有獨特優(yōu)勢,進而推動瘤胃噬菌體相關研究。
本文主要聚焦反芻動物瘤胃生態(tài)系統(tǒng)中的噬菌體群落,并討論瘤胃噬菌體研究方法及現(xiàn)狀,及從其他生境中的噬菌體研究所得到的啟發(fā)。本文還展望了病毒宏基因組學在瘤胃環(huán)境中的應用前景,以期為后續(xù)瘤胃噬菌體組研究提供科學參考。
1 瘤胃噬菌體概述
反芻家畜瘤胃內棲息著數(shù)以萬億計的微生物[12-15](圖1),包括細菌、真菌、原蟲、古生菌和病毒,其中病毒的數(shù)量(>108~109pfu·mL-1)僅次于細菌,且主要為噬菌體(bacteriophage或phage)[16]。瘤胃內的噬菌體具有不同形態(tài),現(xiàn)有研究表明,反芻動物瘤胃中,有尾噬菌體目(Caudovirales)普遍存在,包括肌尾噬菌體科(Myoviridae)、長尾噬菌體科(Siphoviridae)、短尾噬菌體科(Podpviridae)占主導地位[17-18],以及最近被鑒定出來的阿克曼噬菌體科(Ackermannviridae)和赫雷爾噬菌體科(Herelleviridae)[19]。其實,早在20世紀60年代,“瘤胃噬菌體”一詞就已進入人們視野,但對其在瘤胃微生態(tài)系統(tǒng)中的功能知之甚少,直到20世紀80年代末,研究人員才開始對其進行探索[20]。隨著測序技術的發(fā)展,宏病毒組學技術日臻完善,借助該技術手段能夠更好地探索噬菌體這個“暗物質”。已有研究表明,噬菌體在塑造瘤胃菌群結構、維持微生物多樣性和調節(jié)宿主菌代謝等方面發(fā)揮著重要作用[21-22]。對反芻動物瘤胃噬菌體多樣性的研究表明,噬菌體可能是通過裂解宿主細菌從而影響瘤胃細菌種群動態(tài)變化[7,23-24]。Anderson等[23]通過營養(yǎng)調控試驗首次報道了飼糧變化對反芻動物瘤胃噬菌體群落的影響,并表明,噬菌體可通過其編碼的輔助代謝基因(auxiliary metabolic genes, AMGs),經過裂解宿主細菌、產能、復制和微生物代謝的重新編程等一系列過程來影響瘤胃微生態(tài)系統(tǒng)[17,21,23],而其中宿主菌代謝與反芻動物飼料轉化效率密切相關。眾所周知,瘤胃微生物群體結構的穩(wěn)定性和多樣性對于反芻家畜的健康、營養(yǎng)、免疫和生存至關重要,因此,需要重視瘤胃噬菌體在整個瘤胃微生態(tài)系統(tǒng)中的地位、功能及作用。
2 瘤胃噬菌體研究方法
研究噬菌體的傳統(tǒng)方法是顯微鏡觀察和體外分離培養(yǎng),但在實驗室環(huán)境中培養(yǎng)的宿主菌數(shù)量有限;噬菌體還具有形態(tài)小、遺傳進化快等特點,缺乏細菌所擁有的相對保守的系統(tǒng)發(fā)育標記,如16S rRNA基因;且感染不同宿主細菌的噬菌體具有高度特異性,不同噬菌體序列的重疊群非常小[11,25-26]。在瘤胃噬菌體的早期研究中,利用透射電子顯微鏡(transmission electron microscope, TEM)技術,研究人員發(fā)現(xiàn),瘤胃液中存在著形態(tài)各異、高度多樣化的噬菌體種群,并猜測其可能影響瘤胃內細菌的種群[27]。后來,隨著DNA分析技術、脈沖場凝膠電泳技術(pulsed field gel electrophoresis, PFGE)和微生物分離鑒定技術的發(fā)展,研究人員證實,反芻動物瘤胃液中存在大量溶菌性噬菌體[7],并陸續(xù)分離鑒定了多種,包括以壞死梭桿菌(Fusobacteriumnecrophorum)[28]、白色瘤胃球菌(Ruminococcusalbus)[29]、溶纖維丁酸弧菌(Butyrivibriofibrisolvens)[30]和瘤胃擬桿菌(Prevotellaruminicola)[31]等作為宿主菌的噬菌體。TEM技術也隨之被廣泛用于新型噬菌體分離株的形態(tài)學表征[25, 32]。如今,宏基因組學測序技術和生物信息學的快速發(fā)展,使研究人員能更深入地了解病毒群落的結構和功能。宏基因組學(metagenomics)手段是以某一特定環(huán)境樣本中的微生物群體基因組為研究對象,以功能基因篩選和序列測定分析為研究手段,以微生物多樣性、種群結構、進化關系、功能活性、相互作用以及其與環(huán)境間的關系為研究目的的一種新的微生物研究方法[33]。而宏病毒組學(viral metagenomics)則是結合病毒自身特點,將宏基因組學方法應用于病毒學領域[34]。
瘤胃噬菌體宏基因組學研究,主要由病毒樣顆粒(virus-like particles, VLPs)濃縮液制備與生物信息學分析兩部分組成(圖2)。具體步驟包括:1)瘤胃樣本采集,經過濾、消化以及高速離心等技術,去除細菌等其他潛在宿主;2)對樣品進行富集、純化,得到病毒濃縮液;3)根據病毒核酸類型,選擇合適的病毒核酸提取試劑盒或手工提取的方式獲取病毒核酸,構建病毒基因組文庫,并通過高通量測序技術進行宏病毒組測序;4)對測序所得原始數(shù)據進行質控,基于重疊區(qū)(overlap)將高質量測序讀段(reads)拼接為重疊群(contigs);5)通過病毒序列鑒定軟件在重疊群中鑒別、篩選病毒序列,從而了解該環(huán)境中病毒構成;6)對病毒基因組開放閱讀框(open reading frame, ORF)進行預測,再通過注釋工具將 ORF 與多個數(shù)據庫(KEGG、COG、CAZy、CARD等)比對進行功能基因注釋。
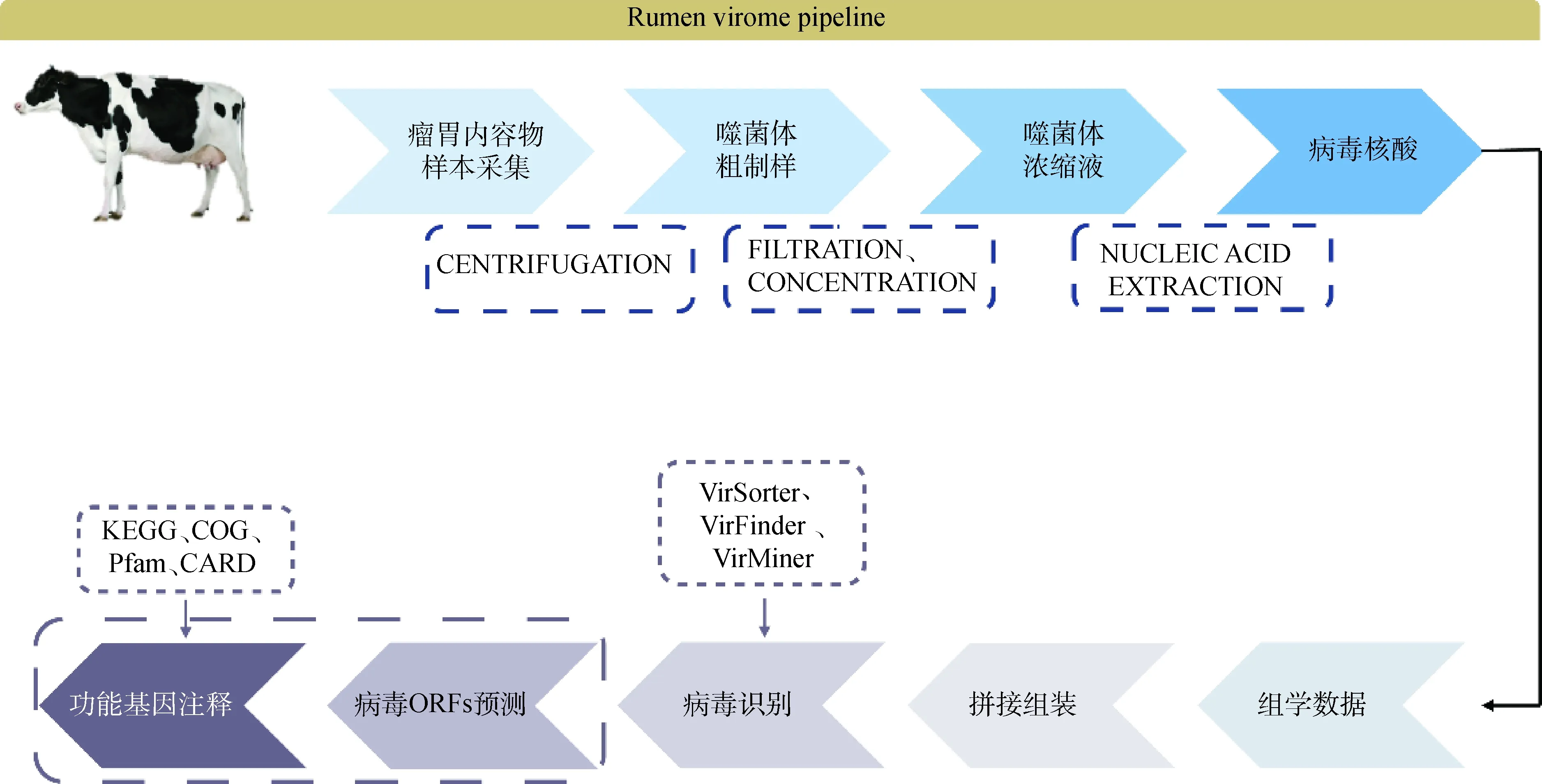
圖2 瘤胃病毒組分析流程圖Fig.2 Flowcharts of rumen virome analysis
2.1 樣品前處理及噬菌體核酸提取
瘤胃噬菌體即為那些以瘤胃細菌為宿主菌的噬菌體,或是那些存在于反芻動物瘤胃體內的噬菌體[31]。由于采樣后,細菌和噬菌體仍然彼此接觸,噬菌體的侵染作用仍持續(xù)存在,長時間孵育會影響噬菌體與微生物比例[35]。因此,樣品采集后要立即超低溫凍存或立即對樣品進行處理,將病毒樣顆粒從樣品中分離并純化,用于病毒遺傳物質提取。現(xiàn)有的提取瘤胃噬菌體核酸的方法大多參考人或小鼠腸道噬菌體及土壤噬菌體分離方法[17,23,31],截至目前,僅有一篇文獻以山羊和綿羊瘤胃液作為瘤胃噬菌體來源,建立了瘤胃噬菌體富集及DNA提取方法(包括過濾、離心、沉淀及核酸提取一系列過程)[18]。
瘤胃環(huán)境異常復雜且微生物多樣性高,瘤胃樣品中噬菌體的分離質量直接關系到病毒宏基因組數(shù)據的質量。Hungate[36]研究表明,大約75%的瘤胃細菌緊密附著在飼料顆粒上,以細菌為宿主的噬菌體也會附著在飼料上,因此,研究瘤胃噬菌體時,需同時采集固相和液相樣品,以便充分囊括瘤胃內的所有噬菌體。在從樣品中分離噬菌體時,常利用噬菌體與其他微生物顆粒大小不同這一特征,借助微孔濾膜、離心等方式來實現(xiàn)噬菌體與其他微生物的分離。但是,實際操作中噬菌體樣極易混入細菌、真菌等微生物,尤其是那些與噬菌體顆粒大小相近的微生物,使得提取的病毒宏基因組中常混有細菌、真菌等其他微生物的核酸[37]。于是,在制備噬菌體粗制樣時,微孔濾膜、化學試劑等的選擇對去除細菌等其他微生物的污染和保證噬菌體群體的代表性具有重要影響,進而對后續(xù)病毒核酸純度、基因功能注釋的準確性產生影響。Concei??o-Neto等[38]研究發(fā)現(xiàn),常見代表性病毒的大小由17到1 000 nm不等,其中,常見噬菌體的大小為125 nm左右。胃腸道病毒顆粒中絕大部分是噬菌體,為了避免細菌的污染,在研究胃腸道噬菌體組時,常選擇孔徑為0.22或0.45 μm的微孔濾膜,當樣本環(huán)境復雜時,為避免遺漏巨型病毒信息,常選擇0.45 μm的微孔濾膜,但為了更有效地防止噬菌體宿主菌污染,0.22 μm的微孔濾膜仍被廣泛結合使用[39]。Friedersdorff等[30]選擇0.45和0.22 μm兩種孔徑的微孔濾膜依次過濾瘤胃液樣品,最終分離得到5種瘤胃溶菌性噬菌體,并對其進行了全基因組學測序。同樣,Namonyo等[18]在建立瘤胃病毒DNA提取方法時也選用了0.45和0.22 μm兩種孔徑的微孔濾膜依次進行過濾。而Klieve等[31]只選擇了孔徑為0.45 μm的微孔濾膜對瘤胃相關樣本進行過濾。雖然現(xiàn)有文獻表明暫未鑒定出巨型瘤胃噬菌體,但并不排除技術手段造成的誤差,為了更全面地研究瘤胃環(huán)境噬菌體群落,研究人員仍需根據研究目的謹慎選擇微孔濾膜的孔徑。
在獲得含噬菌體的粗制樣后,常利用聚乙二醇(polyethylene glycol,PEG)配合不同濃度的NaCl來沉淀噬菌體顆粒以實現(xiàn)噬菌體的濃縮。較為常用的PEG和NaCl用量為25% PEG6000(w/v)和1.0 mol·L-1NaCl[18];10% PEG8000和0.5 mol·L-1NaCl[31]及20% PEG6000和25 mol·L-1NaCl[30]。添加PEG后,常需在低溫下(4 ℃)孵育過夜,以使噬菌體充分沉淀,經過高速離心(13 000×g, 30 min, 4 ℃; 12 000×g, 30 min, 4 ℃或52 350×g, 10 min, 4 ℃)后,倒掉上清液,獲得的沉淀即為噬菌體樣品。根據試驗需求,該樣品可以直接用于后續(xù)試驗,也可經超速離心進一步純化后用于后續(xù)試驗。較為常用的是氯化銫(CsCl)密度梯度離心等,此法可對特定密度范圍內的噬菌體進行純化,并根據密度將VLPs分層。Carroll-Portillo等[40]的測序結果表明,用氯化銫密度梯度離心法富集的VLPs樣品宿主細菌核酸去除率比未經富集的高,但氯化銫密度梯度離心法在富集樣品時,對不同密度的噬菌體有明顯的偏好性。Cordova等[41]也表明,某些噬菌體會由于氯化銫產生的滲透壓脅迫(osmotic shock)而導致噬菌體核酸丟失,因此,研究人員會選擇性采用氯化銫密度梯度離心法。
與瘤胃微生物組研究類似,病毒組研究也需先從分離得到的樣本中提取遺傳物質。鑒于DNA病毒在瘤胃環(huán)境中的優(yōu)勢地位,目前,對于瘤胃噬菌體的研究主要集中于DNA病毒的核酸提取[20]。噬菌體核酸提取主要有兩種方式:第一種是試劑盒法,如Roche high pure viral nucleic acid large volume kit (Roche, Switzerland)[18]、QIAamp Ultra Sens Virus Kit (Qiagen)[23]和Fast DNATMSpin Kit for Soil (MP Biomedicals, Solon, OH, United States)[31]等,這類試劑盒可用于DNA病毒核酸的提取,試劑盒具有操作方便、提取純度高且不需要直接接觸有害化學試劑等優(yōu)點;第二種是傳統(tǒng)的苯酚-氯仿提取法,利用核酸、蛋白質等雜質在水相和有機相中溶解性不同而重新分配的性質來實現(xiàn)核酸提取,該方法價格低廉,但耗時長且提取濃度低,常需進一步純化后才能達到測序要求[42]。試驗中,若遇提取的噬菌體DNA濃度過低而不能滿足測序要求的情況,可利用多重置換擴增(multiple displacement amplification, MDA)對核酸進行擴增,得到的DNA可用于構建基因文庫或高通量測序[43],但擴增技術所得到的產物可能存在偏好性,還是建議提高原始噬菌體樣品富集量以保證結果的準確性[3]。
2.2 高通量測序與病毒序列鑒定
高通量測序技術(high-throughput sequencing, HTS) 是對傳統(tǒng)Sanger測序技術革命性的變革,可對數(shù)百萬個DNA分子進行同時測序,并可深入地對一個物種的基因組和轉錄組進行整體分析,因此,也稱其為下一代測序技術 (next generation sequencing, NGS)[44]。HTS具有通量高、速度快、成本低等優(yōu)點,已廣泛應用于宏病毒組學的研究中。近年來,牛津納米孔技術(Oxford nanopore technology, ONT)作為新興的單分子實時測序技術(single molecule real-time sequencing technology, SMRT)之一,具有快速制備文庫,超長讀取和實時數(shù)據采集等優(yōu)勢[45]。該技術的核心是蛋白質納米孔,通過產生覆蓋單個病毒顆粒內所有突變的基因組長度讀數(shù)來獲得病毒基因組。在此基礎上,中國科學院微生物研究所王軍研究員團隊開發(fā)了一種新的工作流程(包括病毒顆粒富集、核酸的逆轉錄和擴增以及生物信息學分析),并首次使用ONT PromethlON平臺對人類病毒組進行表征[46]。相較于傳統(tǒng)的二代測序,ONT具有讀長長的優(yōu)點,因此,無需進行PCR擴增,便可以在更短時間內、更準確地從樣品中生成數(shù)據。
高通量測序后首先要對原始數(shù)據進行質量控制和過濾,去除宿主核酸,再進行序列的拼接,并運用專業(yè)軟件來鑒定病毒序列,確定該序列的生物群落來源,篩選出有用的基因信息。對噬菌體基因組進行組裝時,一般挑選1 000 bp以上組裝序列,再應用一系列軟件對病毒contig進行鑒定。表1列出了常用于噬菌體鑒定的生物信息學分析工具。
從同時包含噬菌體及其宿主的混合基因組數(shù)據集中準確識別噬菌體,是分析樣品中噬菌體組成的關鍵步驟。目前,有非常多基于序列或基于重疊群對病毒進行鑒定的生物信息學分析軟件。VirSorter(https://github.com/simroux/VirSorter)可利用概率模型和大量病毒數(shù)據最大限度地檢測新病毒,雖然該工具在很大程度上依賴對已有病毒基因組的相似性搜索,但它有一個顯著優(yōu)勢,即可手動管理病毒參考基因組數(shù)據庫,并補充了以海水、人體腸道、肺部組織及唾液樣本為來源的宏病毒組序列[47]。2017年,Ren等[48]開發(fā)的VirFinder(https://github.com/jessieren/VirFinder)是通過利用細菌和病毒在K-mer上的差異,將病毒從宏基因組數(shù)據集中識別出來,與VirSorter相比,它能鑒定出更多潛在病毒,尤其是大小在1 000 bp以下的病毒contig。但VirSorter和VirFinder進行噬菌體分析的功能仍相對有限,在鑒定噬菌體重疊群后,無法預測噬菌體與宿主間的相互作用。MARVEL(https://github.com/LaboratorioBioinformatica/MARVEL)是2018年由Amgarten等[49]開發(fā)的工具,以其對Caudovirales目dsDNA病毒的有效注釋能力而為人所知,但該工具鑒定RNA病毒的錯報率非常高。2019年開發(fā)的VirMiner(https://github.com/TingtZHENG/VirMiner)則針對噬菌體分析功能做出了改進,可通過預測模型從宏基因組數(shù)據集中預測噬菌體重疊群并進行下游分析,包括功能基因注釋及噬菌體與宿主間關系預測等[50]。VIBRANT(https://github.com/AnantharamanLab/VIBRANT),是2020年開發(fā)的利用迭代注釋進行病毒識別的工具,它能高效識別以細菌為宿主的多種病毒(包括dsDNA、ssDNA、dsRNA和ssRNA病毒),并從宿主序列中提取前病毒區(qū)域,從而預測到其他分析工具無法預測到的前病毒(prophage),且能解析環(huán)境中不同病毒之間新陳代謝的能力[51]。2021年,Guo等[52]改進并發(fā)布了VirSorter2(https://github.com/jiarong/VirSorter2),但VirSorter仍是最被廣泛使用的工具。現(xiàn)有的每種工具都有其局限性,應根據實際需求選擇使用或聯(lián)合使用不同的軟件。
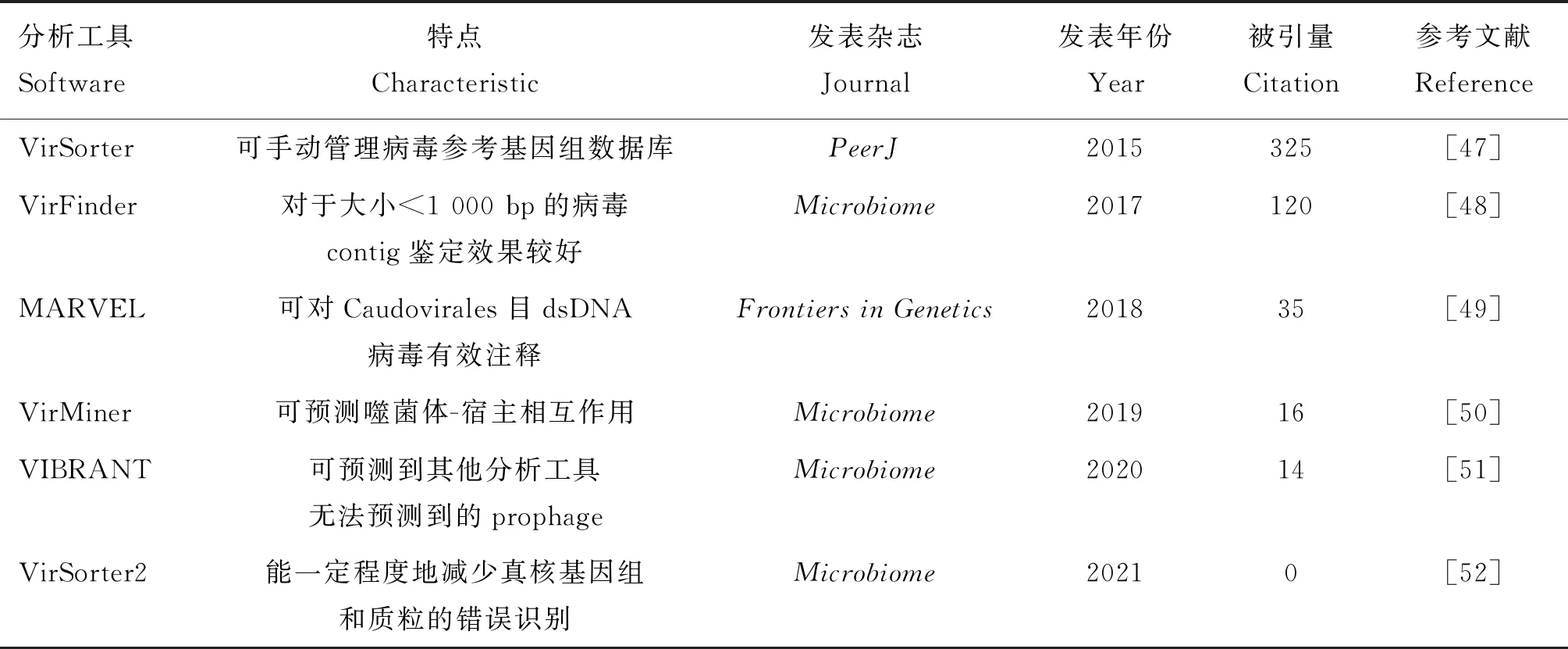
表1 病毒鑒定工具
2.3 病毒功能基因注釋
使用BLAST軟件進行基因預測,并將獲得的非冗余基因組與RefSeq病毒數(shù)據庫的參考序列進行比對,獲取該基因的功能信息[53]。通過對病毒功能基因注釋,能夠對深入認識病毒個體生命過程提供理論基礎,還有助于了解病毒群落的生態(tài)過程及與宿主群落的互作網絡,從而闡釋病毒與宿主間復雜的相互作用機制。
京都基因與基因組百科全書(Kyoto encyclopedia of genes and genomes,KEGG)是一個系統(tǒng)分析基因功能的知識庫,KEGG具有強大的圖形功能,可利用圖形來介紹眾多代謝途徑(包括各種代謝通路、合成通路、膜轉運、信號傳遞、細胞周期以及疾病相關通路等)以及各個途徑間的關系,這樣可以直觀地反映基因與相關代謝的關系[54]。蛋白質直系同源簇(clusters of orthologous groups of proteins,COG)數(shù)據庫,可將不同物種中的直系同源基因進行聚類,通過在不同物種中建立相關同源蛋白簇來預知位置蛋白質的功能,同時也為分子系統(tǒng)發(fā)育分析提供數(shù)據基礎。2015年,Galperin等[55]基于完整噬菌體基因組中的編碼蛋白系統(tǒng)進化關系構建了 POG (phage orthologous groups)數(shù)據庫,可在雙鏈DNA噬菌體的全基因組序列中鑒定出保守的直系同源簇。Pfam 數(shù)據庫是蛋白質家族的數(shù)據庫,根據多序列比對結果和隱馬爾可夫模型(hidden markov model, HMMs),可查詢蛋白質家族或蛋白結構域的注釋、結構及多序列比對信息,被廣泛用于基因功能注釋[56]。抗生素的濫用對畜牧業(yè)造成了深遠影響,雖然抗生素對病毒并無直接作用,但有研究發(fā)現(xiàn),瘤胃噬菌體攜帶抗生素耐藥性基因[23],因此,對病毒功能基因序列進行耐藥性檢測就顯得尤為重要。抗生素綜合研究數(shù)據庫(comprehensive antibiotic resistance database,CARD) 可在細菌耐藥性的分子基礎上,提供參考 DNA 和蛋白質序列、檢測模型和生物信息學工具,通過與該數(shù)據庫進行比對,可用于關聯(lián)抗生素模塊及其目標、抗性機制、基因變異等信息與耐藥基因相關的注釋信息[57]。
3 瘤胃噬菌體基因組研究趨勢及進展
借助Web of Science核心合集以“rumen bacteriophage”、“rumen phage”或“rumen virus”為關鍵詞進行檢索,2000—2021年時間段內共有108篇相關文獻,且發(fā)文量呈現(xiàn)整體上升趨勢(圖3a),2021年發(fā)表的5篇文獻并未在圖中標明。其中,美國、澳大利亞、印度、新西蘭和加拿大的發(fā)文量分別為33.66%、15.39%、9.62%、8.65%和7.69%,而我國在瘤胃噬菌體這一領域的研究嚴重滯后。所發(fā)表的關于瘤胃噬菌體的文章中,基于宏基因組學的文獻僅有7篇[17-18,21,23-24,58-59],涉及研究動物包括水牛、奶牛、綿羊、山羊和麋鹿,且很少涉及瘤胃噬菌體群落動態(tài)變化規(guī)律及瘤胃噬菌體在整個瘤胃微生態(tài)系統(tǒng)中的作用。隨后,利用VOSviewer進行關鍵詞密度聚類,并進行可視化分析[60](圖3b),圖中結點越接近黃色,表示此為研究熱點,可發(fā)現(xiàn)瘤胃環(huán)境病毒研究熱點為噬菌體及宏基因組層面。而以“gut phage”或“gut virus”作為關鍵詞檢索,在2000—2021年時間段內共有4 449篇相關文獻,這說明,瘤胃噬菌體是一個極具發(fā)掘空間的研究領域。
對病毒進行全基因組測序(complete genome sequencing),又叫單病毒基因組學(single-virus genomics, SVG),可獲得特定病毒(噬菌體)的全部核酸序列。SVG即以純培養(yǎng)的病毒為研究對象,進行核酸提取、文庫構建并對單個病毒進行全基因組測序,SVG從頭組裝可以規(guī)避病毒遺傳物質多樣性的問題,從而構建更完整的病毒基因組,以獲得更全面的病毒數(shù)據庫的信息[61]。目前,僅有兩篇基于SVG來研究瘤胃噬菌體的文章,表2綜述了噬菌體及其宿主信息。
Friedersdorff等[30]以瘤胃擬桿菌(Prevotellaruminicola)、白色瘤胃球菌(Streptococcusbovis)及瘤胃鏈球菌(Ruminococcusalbus)為宿主,采用雙層平板法(plaque assay)從瘤胃相關樣品(廢水、牛糞及瘤胃液)中分離出5株烈性噬菌體,這是首次對瘤胃環(huán)境中以特定細菌為宿主的噬菌體的基因組研究。隨后對這5株噬菌體進行全基因組測序,并通過 Glimmer和Prodigal在線工具對噬菌體全基因組序列進行潛在編碼序列(coding sequence, CDS)預測和功能注釋。發(fā)現(xiàn)在φBrb01和φBrb02基因組中,CDS 可分為DNA 復制轉錄(DNA replication and transcription)、DNA 包裝(DNA packaging)及宿主裂解(Host lysis)3個功能組。溶纖維丁酸弧菌(Butyrivibriofibrisolvens)是瘤胃內的一種優(yōu)勢菌,對纖維降解和蛋白質分解等有重要作用。Friedersdorff等[30]以其為宿主,從瘤胃液及糞便中分離了5株噬菌體,并進行全基因組測序和利用 Glimmer和GeneMarkS在線工具進行功能注釋。結果顯示,在科水平上,瘤胃噬菌體以有尾噬菌體為主,其中長尾噬菌體科占主導地位,與前人研究結果一致[17-18]。Ceridwen屬噬菌體基因組中與其他噬菌體基因組同源的ORF中,其中20個與Siphoviridae科噬菌體有同源性,1個與Podoviridae科噬菌體有同源性。另外,用PHACTS軟件對噬菌體生活史進行預測發(fā)現(xiàn),這10株噬菌體中僅有Arawn和Idris屬噬菌體基因組中存在編碼整合酶的基因,表明這兩株噬菌體可能為溶源性噬菌體(lysogenic phage),其他8株噬菌體為溶菌性噬菌體(lytic phage)。值得注意的一點是,從瘤胃液中分離出的Bo-Finn噬菌體與從牛糞便中分離出的Arian噬菌體基因組序列相似性可達98.6%,它們屬于同種噬菌體。
4 不同生境噬菌體研究進展
自2002年美國學者Breitbart等[62]首次對海洋病毒群(virome)進行宏基因組測序,到2020年,全球范圍內的研究已報道了近20萬個病毒種群[63]。目前,噬菌體的研究已然成為研究不同生境內微生物群落結構和功能的焦點,如哺乳動物腸道噬菌體與機體健康和疾病發(fā)生已受到廣泛關注。關于不同生境噬菌體(如人體腸道、土壤等)的研究已證實,噬菌體是導致環(huán)境中宿主細菌死亡的主要原因[64],且噬菌體可作為水平轉移基因(horizontal gene trans-fer, HGT)的重要載體,與宿主菌進行遺傳物質的交換,進而調節(jié)細菌的進化和多樣性;值得注意的是,HGT也是導致抗生素抗性基因(antibiotic resistance genes, ARGs)擴散的重要因素[65]。鑒于瘤胃內噬菌體研究相對滯后的現(xiàn)狀,可以適度借鑒其他環(huán)境樣本中的噬菌體研究方法并用于瘤胃噬菌體研究。

圖3 瘤胃噬菌體領域年發(fā)表論文數(shù)量(a)及關鍵詞密度圖譜(b)Fig.3 Number of publications(a) and keyword dendity network map(b) in the field of rumen virus (bacteriophage)
許多環(huán)境噬菌體的研究得益于微生物多組學方法聯(lián)用[66-67],取得了喜人的成果。例如,Emerson等[66]利用宏基因組和宏轉錄組揭示了氣候變化中噬菌體對復雜碳降解的潛能。此外,Brum等[67]結合蛋白質組學和宏基因組學,從海洋環(huán)境中鑒定出多樣性極高的病毒衣殼蛋白。類似的,蛋白質組學這一新興領域在瘤胃微生態(tài)系統(tǒng)中的研究表明,瘤胃噬菌體介導的細胞裂解會釋放微生物胞內酶,包括那些參與碳水化合物分解的微生物酶,這些酶的釋放會促進瘤胃內飼料的降解[59]。
除了以上能通過組學技術挖掘出的數(shù)據外,病毒宏基因組測序結果中大部分序列為未知序列,為了充分利用這些未知數(shù)據,Brum等[67]通過對預測的蛋白質序列進行全長相似性聚類,揭示了七大洋海洋樣本中存在一個核心病毒基因集,這樣可以從未知序列中得到另一種“已知”結果。Swain等[22]考慮到瘤胃病毒在不同個體間存在的差異,運用類似方法發(fā)現(xiàn)了核心瘤胃病毒基因集。此外,Allen等[68]研究海洋微生物群落時采用了一種基于噬菌體基因組標簽的方法(phage genome signature-based recovery, PGSR),從宏基因組中提取噬菌體序列,并結合病毒/細菌比(virus-to-bacteria ratio,VBR)來評估病毒對細菌群落的影響,該方法有助于研究人員了解噬菌體在塑造整個海洋微生物群落結構中的作用。但目前未見此法運用于瘤胃噬菌體研究。
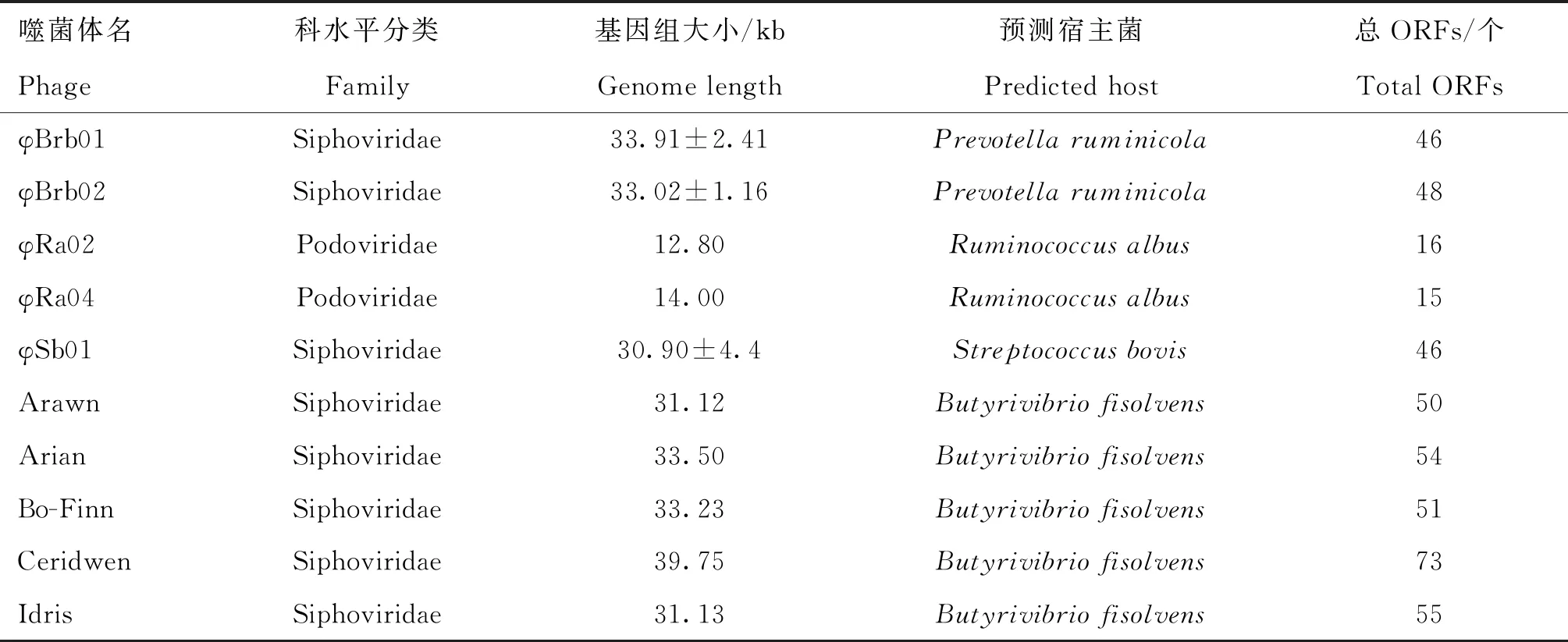
表2 已報道的獲得全基因組信息的瘤胃噬菌體
值得注意的是,不同生態(tài)環(huán)境中研究方法的適用性有限。即使兩個生態(tài)系統(tǒng)具有非常相似的病毒群落結構,他們潛在的微生態(tài)關系也不盡相同。比如,極地水體環(huán)境噬菌體和胃腸道環(huán)境噬菌體中,溶源性噬菌體較溶菌性噬菌體在數(shù)量上占有優(yōu)勢[69-71]。但極地海洋生態(tài)系統(tǒng)中,隨著細菌豐度增加,溶源性噬菌體會從溶源循環(huán)(lysogenic cycle)轉變?yōu)槿芫h(huán)(lytic cycle)[69];而在腸道中,宿主菌豐度增加時,溶源性噬菌體會持續(xù)占領主導地位[70-71]。因此,在將不同生境中噬菌體研究方法及生物學概念外推到瘤胃環(huán)境時仍需謹慎。
5 瘤胃宏病毒組的前景與展望
隨著宏基因組學和生物信息學的發(fā)展,研究人員可以不再需要體外分離培養(yǎng)噬菌體,就能獲得一些分散、豐度低的病毒的結構和功能信息,也逐步增進對瘤胃內噬菌體的多樣性及潛在功能的認知。但是,現(xiàn)階段學術界對瘤胃噬菌體及其功能基因的科學認知仍然十分有限,筆者結合自身體會,認為今后此領域的研究應主要聚焦在以下幾個方面:
5.1宏基因組學研究中,瘤胃噬菌體樣品制備時主要通過微孔濾膜過濾的方式進行純化富集,因此,難免會因為濾膜孔徑選擇過小而造成顆粒較大的噬菌體的遺傳信息的遺漏。現(xiàn)階段瘤胃噬菌體的提取、宏病毒組分析等缺乏統(tǒng)一技術規(guī)范,建立成熟的噬菌體分離純化實驗室標準化操作流程以及針對巨型噬菌體的優(yōu)化提取和富集技術是當務之急。
5.2目前的研究主要聚焦在瘤胃DNA病毒,對于RNA病毒的研究較少。未來要利用多組學手段(如宏基因組、宏蛋白組、宏轉錄組和宏代謝組等)結合生物信息學技術,同步關注瘤胃RNA病毒的基因功能,盡可能獲得全面的噬菌體信息,這有助于探明瘤胃整體病毒的生態(tài)功能和作用機制。
5.3鑒于噬菌體種群在個體間差異顯著且噬菌體種群存在于反芻動物整個消化道,需進一步研究不同生理階段、胃腸道不同部位噬菌體群落對微生物群落的影響。
5.4盡管病毒宏基因組學為研究人員提供了一種不需分離培養(yǎng)即可了解環(huán)境中全部病毒基因組信息的方法,但仍需要以瘤胃優(yōu)勢菌群為宿主菌,對噬菌體進行分離培養(yǎng),這將有助于全面了解噬菌體的生物學特性以及它們在維持瘤胃動態(tài)平衡中的作用。
5.5關于瘤胃病毒的研究在國內處于亟待開發(fā)的階段,瘤胃是一個復雜、多變的微生態(tài)系統(tǒng),蘊藏著豐富的基因資源,有大量新病毒等待研究人員去挖掘。故需要大力發(fā)展全基因組擴增技術和測序技術,不斷完善動物胃腸道病毒資源庫,豐富瘤胃噬菌體基因組數(shù)據庫,揭示噬菌體塑造瘤胃細菌群體結構的機制。
5.6我國于2020年開始對飼料全面禁抗,鑒于瘤胃噬菌體對瘤胃微生物的裂解作用及對瘤胃營養(yǎng)物質循環(huán)的影響,噬菌體制劑作為抗生素替代品將成為畜牧界的重大課題。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車工程學報(2017年2期)2017-07-05 08:13:02
國際商務財會(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
