POE-g-GMA對PLA/PP共混體系結構及性能的影響
2022-03-03 03:20:22謝永健陳鑫亮樊炳宇
石油化工 2022年1期
高 尚,謝永健,陳鑫亮,楊 利,樊炳宇,王 平
(安徽建筑大學 材料與化學工程學院 安徽省先進建筑材料國際聯合研究中心,安徽 合肥 230601)
聚乳酸(PLA)因具有優異的機械性能和生物可降解性能,以及能用作石油基聚合物替代品[1-4]等特點,受到廣泛的關注,但由于制品較脆等因素,限制了PLA基材料的廣泛應用。通過共混改性是提高PLA綜合性能的方法之一[5-8]。聚丙烯(PP)具有優良的綜合性能,將其與PLA共混能夠提高PLA的綜合性能。但PLA與PP較弱的熱力學相容性導致PLA/PP共混材料的性能較差。目前,添加交聯劑或相容劑改善兩相之間的界面狀態或界面結構是提高PLA與PP相容性的主要方法之一,如苯乙烯-(乙烯-丁烯)-苯乙烯嵌段共聚物、乙烯-丙烯酸甲酯-甲基丙烯酸縮水甘油酯無規三元共聚物、過氧化二異丙苯、聚二甲基硅氧烷-聚乙二醇彈性粒子等[9-12]。Zawawi等[13]利用PP接枝馬來酸酐以改善不同相之間的界面狀態,并加入蒙脫石有機納米黏土增強材料的機械性能。Savas等[14]利用乙烯-丙烯酸丁酯及碳纖維增強PLA/PP體系的相容性和機械性能,制備了強度更高的可再生復合材料。關于制備具有良好剛韌平衡性的PLA/PP共混復合材料的研究較多,但對PLA/PP共混復合體系界面形貌演變的過程和機理的研究報道較少。
本工作采用甲基丙烯酸縮水甘油酯(GMA)接枝乙烯-辛烯共聚物(POE)得到的POE-g-GMA作為相容劑引入到PLA/PP共混體系中,利用FTIR,DSC,SEM等方法研究了POE-g-GMA對PLA/PP共混體系加工性能、流變性能、結晶性能和機械性能的影響,并對POE-g-GMA的增容機理進行了探討。
1 實驗部分
1.1 實驗原料
PLA:牌號4032D,Nature Works公司,Mw=195 000;PP:牌號JH380,Lotte Chemical公司,Mw=220 000;POE-g-GMA:牌號SOG-02,佳易容聚合物有限公司,GMA含量約2%(w)。
1.2 實驗儀器
RM-200C型轉矩流變儀:哈爾濱哈普電氣技術有限責任公司;XH-406C型抽真空平板硫化儀:東莞市錫華檢測儀器有限公司;WZY-4030型智能數控萬能制樣機:承德金和儀器制造有限公司;CMT4304型微機控制電子萬能試驗機:美特斯工業系統有限公司;Nicolet6700型傅里葉變換紅外光譜儀:賽默飛世爾公司;DSC 30型差示掃描量熱儀:上海精科天美貿易有限公司;DHR-2型旋轉流變儀:TA公司;JSM-6490LV型鎢燈絲掃描電子顯微鏡:日本電子制造株式會社。
1.3 試樣制備
首先將所有原料在真空干燥箱中60 ℃下干燥24 h,然后按表1中的比例加入到轉矩流變儀中,在180 ℃、轉速50 r/mim條件下,熔融共混6 min。將所得材料充分冷卻干燥后置于真空平板硫化儀中在180 ℃,10 MPa下,熱壓5 min再冷壓3 min成型,并參考文獻[15]的標準將材料裁成啞鈴形樣條。
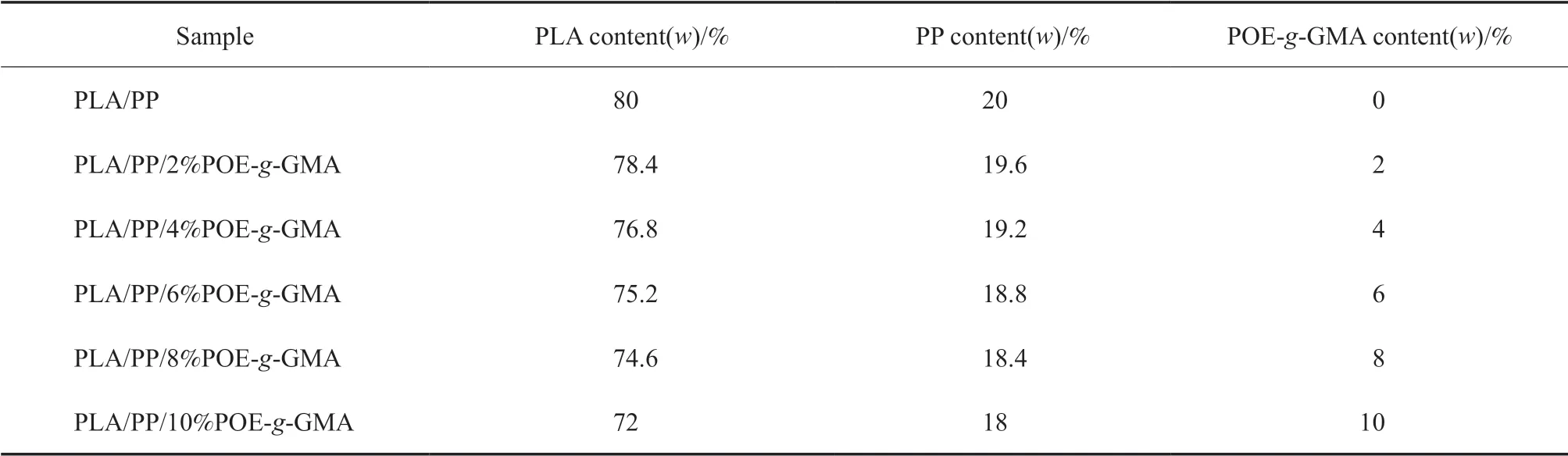
表1 PLA/POE-g-GMA/PP共混物的組成與配比Table 1 Composition of PLA/POE-g-GMA/PP blends
1.4 測試與表征
采用傅立葉變換紅外光譜儀研究POE-g-GMA對PLA/PP共混體系分子間相互作用的影響:在衰減全反射模式下,掃描波長范圍為600~4 000 cm-1,掃描分辨率為4 cm-1,掃描次數為32次。
采用旋轉流變儀表征試樣的流變行為:通過萬能制樣機將試樣制成直徑為25 mm,厚度為0.8 mm的圓片,然后將圓片置于氮氣流量為5 L/min的平行板旋轉流變儀的平行板夾具中,然后升至180 ℃將試樣熔融,恒溫5 min消除熱歷史,并進行動態頻率掃描,動態掃描條件為:固定應變1%,掃描角頻率范圍0.04~400 rad/s,進行小振幅振蕩剪切,測試動態儲能模量(G')、動態損耗模量(G'')和復數黏度(η*)隨掃描角頻率的變化關系。
采用差示掃描量熱儀研究POE-g-GMA對PLA/PP共混體系凝聚態結構的影響:取10 mg左右試樣,在氮氣氣氛下,以10 ℃/min升溫速率升至200 ℃,并在200 ℃下等溫5 min以消除熱歷史,然后以10 ℃/min的降溫速率降至-30 ℃,最終以10 ℃/min的升溫速率升至200 ℃進行二次升溫。
采用微機控制電子萬能試驗機研究POE-g-GMA對PLA/PP共混體系機械性能的影響,通過萬能制樣機制成啞鈴形樣條,并以5 mm/min的拉伸速率進行拉伸測試。
采用掃描電子顯微鏡在加速電壓5 kV下,觀察PLA/PP/POE-g-GMA共混試樣的拉伸斷面。
2 結果與討論
2.1 POE-g-GMA對PLA/PP共混體系加工性能的影響
圖1為PLA/POE-g-GMA/PP共混體系的扭矩-時間曲線。
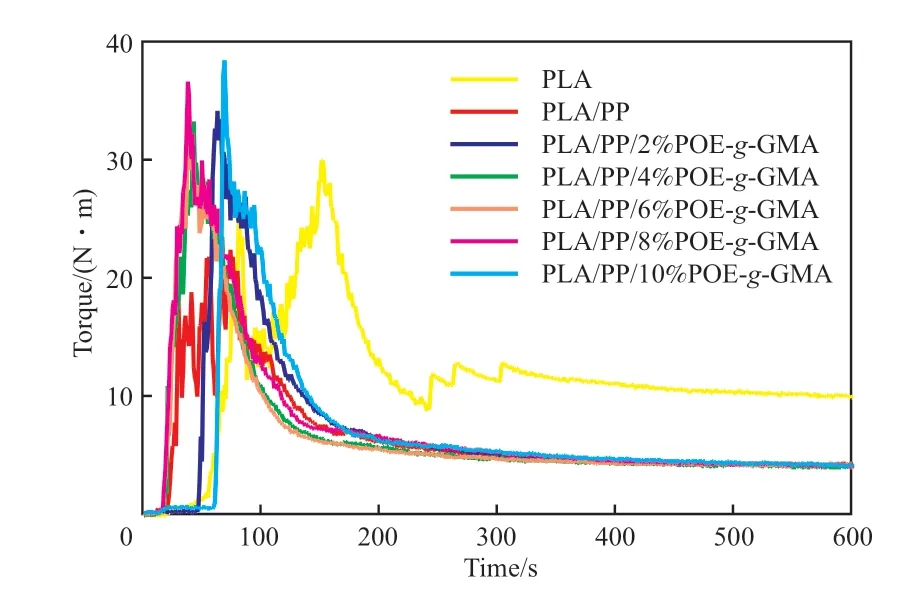
圖1 PLA和PLA/POE-g-GMA/PP共混物的扭矩-時間關系曲線Fig.1 The torque-time relationship diagram of PLA and PLA/POE-g-GMA/PP blends.
由圖1可知,PLA的平衡扭矩為9.8 N·m,添加20%(w)的PP后,共混物熔體強度降低,平衡扭矩降至4.2 N·m。在此基礎上,進一步向PLA/PP共混體系中引入POE-g-GMA,發現隨著POE-g-GMA含量的增加,PLA/POE-g-GMA/PP共混體系的平衡扭矩呈先減小后增加的趨勢。原因為共混體系中分子量相對較小的POE-g-GMA能夠通過增塑作用降低共混體系的熔體強度,但同時POE-g-GMA中的環氧基團能夠與PLA的端羥基和羧基通過擴鏈反應增加熔體強度,在兩者的共同作用下,平衡扭矩保持相對穩定。
2.2 POE-g-GMA對PLA/PP共混體系分子間相互作用的影響
圖2為試樣的FTIR譜圖。從圖2可看出,PLA在1 180,1 750,2 951,2 999 cm-1處出現特征峰,分別對應C—O—C、C=O、—CH、—CH3的伸縮振動。共混物PLA/PP與PLA的譜圖基本相同,表明PLA與PP共混后基團未發生明顯變化。在加入相容劑POE-g-GMA后,試樣在2 918,2 839 cm-1處出現了POE-g-GMA的特征吸收峰,且各峰面積發生明顯變化,表明POE-g-GMA的GMA基團與PLA的端羥基或羧基在共混過程中發生了化學反應。
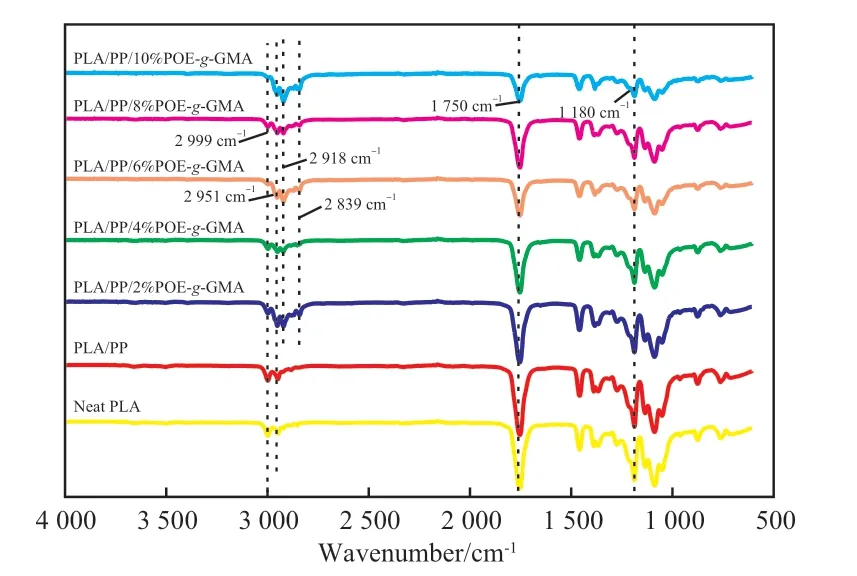
圖2 試樣的FTIR譜圖Fig.2 FTIR spectra of samples.
2.3 POE-g-GMA對PLA/PP共混體系流變性能的影響
POE-g-GMA對PLA/PP共混體系流變性能的影響見圖3。從圖3a可看出,PLA和PLA/POEg-GMA/PP共混體系的G'隨角頻率的增大而逐漸增大,主要表現為彈性行為[16]。在低頻區,G'隨著PP的加入和POE-g-GMA含量的增加而逐漸增加,說明共混物中的分子鏈的纏結程度增加。這是因為,PP的分子鏈較PLA鏈更具彈性,因此更容易纏結,分子鏈纏結密度提高,導致熔體的可逆彈性變形更高;同時,POE-g-GMA的環氧基團與PLA的端羥基和羧基的反應增加了PLA與PP間的相互作用,從而導致較高的彈性響應。從圖3b可看出,POE-g-GMA的引入導致G''逐漸增加,進一步表明了分子鏈間纏結密度和相互作用增強,導致內耗增加。從圖3c可看出,PLA,PP,PLA/PP和PLA/2%POE-g-GMA/PP在低頻區的復數黏度變化不大,呈牛頓特性,隨后在高頻時呈復數黏度降低的非牛頓特性。當POE-g-GMA含量超過2%(w)后,在整個頻率范圍內,PLA/POE-g-GMA/PP的復數黏度高于PLA,且隨角頻率的增加而降低,最終趨于平穩[17],即在整個頻率范圍內均呈非牛頓特性。產生上述現象是因為POE-g-GMA增強了PLA和PP之間相互作用。從圖3d可看出,Tanδ(G''/G')表示動態條件下能量損失與能量存儲的比值。隨POE-g-GMA含量的增加,試樣的Tanδ逐漸降低,表明POE-g-GMA改善了PLA與PP的相互作用,共混物的熔融行為得到了增強。
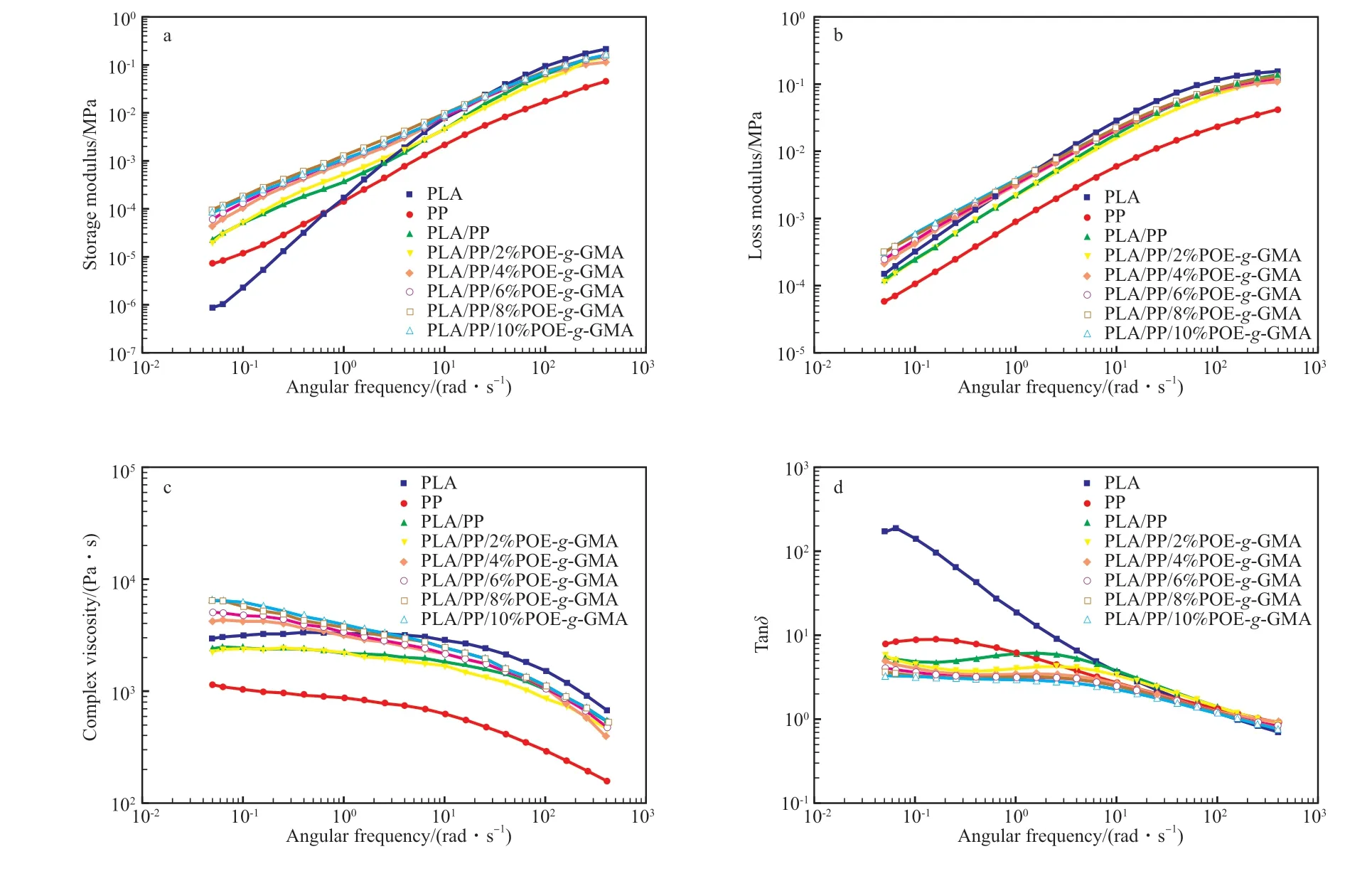
圖3 POE-g-GMA對PLA/PP共混體系流變性能的影響Fig.3 Effect of POE-g-GMA on the rheological properties of PLA/PP blends.a Storage modulus;b Loss modulus;c Complex viscosity;d Tanδ
2.4 POE-g-GMA對PLA/PP共混體系結晶性能的影響
圖4 為試樣的DSC曲線。由圖4可知,PLA的玻璃化轉變溫度(Tg)為60.6 ℃,冷結晶溫度(Tcc)為110.6 ℃,結晶度為4%,結晶能力較弱。與PP共混后,PLA/PP相比PLA,Tg變化不大,但Tcc降低,即PLA/PP共混物的結晶能力增強。隨POE-g-GMA用量的增大,PLA/POE-g-GMA/PP的Tg與Tcc呈升高趨勢,結晶度則先升高后降低。6%(w)的POE-g-GMA即可使共混材料PLA/POEg-GMA/PP的結晶度提升至30.4%,但當POE-g-GMA添加量為10%(w)時,PLA/POE-g-GMA/PP的Tcc升至97.6 ℃,結晶度降至20.7%。原因是低含量的POE-g-GMA具有增塑作用,可提高PLA鏈段的運動能力,從而提高材料的結晶度;而POE-g-GMA含量過高時,環氧基團與PLA端基反應程度提高,導致在共混體系中形成微交聯結構,反而限制了PLA的鏈段運動,使PLA的結晶能力下降。此外,與PLA/PP相比,PLA/POE-g-GMA/PP的熔融溫度升高,說明PLA的晶體更加完善。
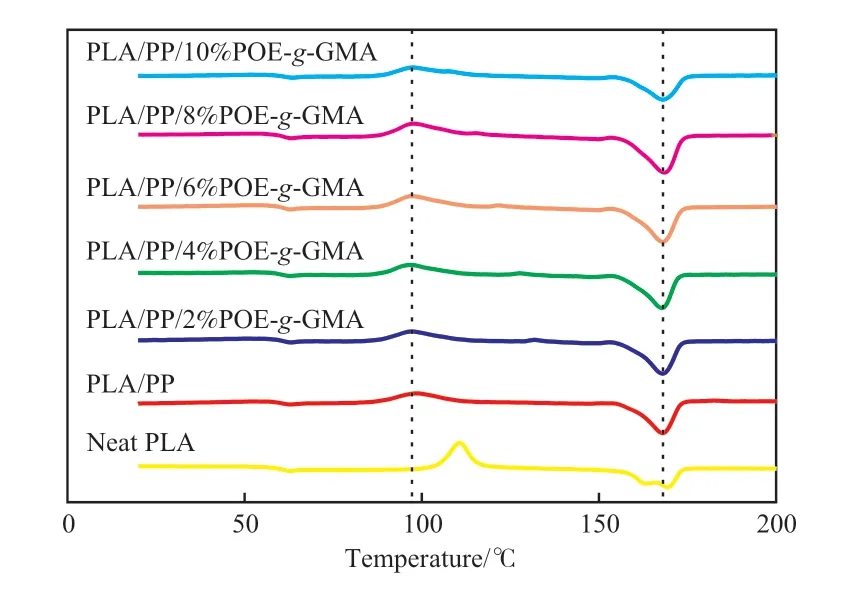
圖4 PLA和PLA/POE-g-GMA/PP共混物的DSC二次升溫曲線Fig.4 DSC secondary heating curve of PLA and PLA/POE-g-GMA/PP blends.
2.5 POE-g-GMA對PLA/PP共混體系機械性能的影響
圖5為試樣的力學性能。由圖5可知,PLA的拉伸強度為59.9 MPa,但斷裂伸長率僅為3.4%,呈明顯的脆性。與PP共混后,PLA/PP的拉伸強度與斷裂伸長率均下降,說明PP與PLA之間較差的相容性導致PP無法對PLA進行有效增韌。引入POE-g-GMA后,PLA/POE-g-GMA/PP的拉伸強度減小,而斷裂伸長率增加。當POE-g-GMA含量為6%(w)時,PLA/POE-g-GMA/PP的斷裂伸長率由2.5%提高至25%,較PLA/PP提高了10倍,且拉伸強度達到29.2 MPa,材料的剛韌平衡性良好。當POE-g-GMA的含量超過6%(w)時,共混材料的斷裂伸長率反而下降,這是因為過量的POE-g-GMA會在體系中形成團聚,降低鏈段運動能力,受到外力作用時難以通過鏈段運動的緩沖而吸收能量,同時POE-g-GMA自身的團聚會產生應力集中,使材料更容易斷裂。
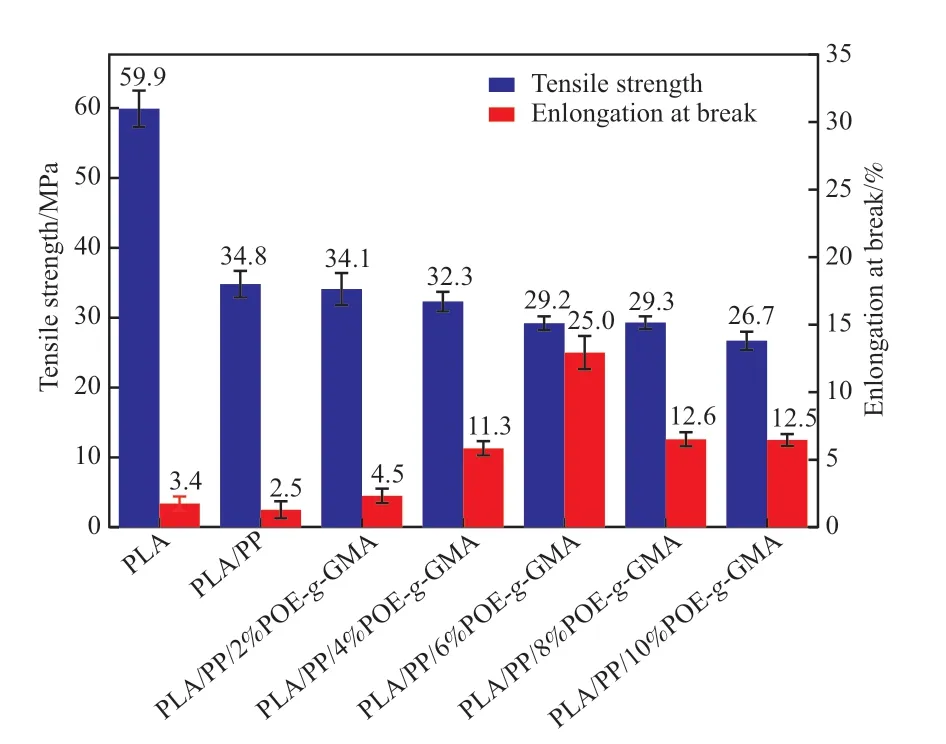
圖5 PLA/POE-g-GMA/PP共混體系的力學性能Fig.5 The mechanical properties of PLA/POE-g-GMA/PP blends.
2.6 POE-g-GMA對PLA/PP共混體系界面狀態的影響
圖6 為試樣拉伸斷裂面的SEM照片。從圖6可看出,PLA斷面光滑,為典型的脆性斷裂。PLA/PP則呈典型的“海-島”結構,存在較大尺寸的顆粒及空洞,表明PLA與PP界面相互作用較弱。引入POE-g-GMA后,PP相的尺寸明顯減小,且分散均勻,表明PLA與PP相容性有所提高。當POE-g-GMA含量為6%(w)時,PLA與PP的相界面模糊,并發現類似纖維狀的結構,表明PLA和PP的相容性較好,界面相互作用較強。POEg-GMA的加入對界面狀態的改善機理見圖7。從圖7可看出,POE-g-GMA中的環氧基團與PLA端基反應,形成微交聯結構,使兩相結合得更加緊密。繼續增加POE-g-GMA的用量,發現共混物相結構由“海-島”結構向連續相結構轉變,但在連續相中發現PP顆粒拔出現象,這是因為過量的POE-g-GMA聚集,導致材料內部發生應力集中降低了共混材料的力學性能。

圖6 試樣拉伸斷裂面的SEM照片Fig.6 SEM image of tensile fracture surface of samples.
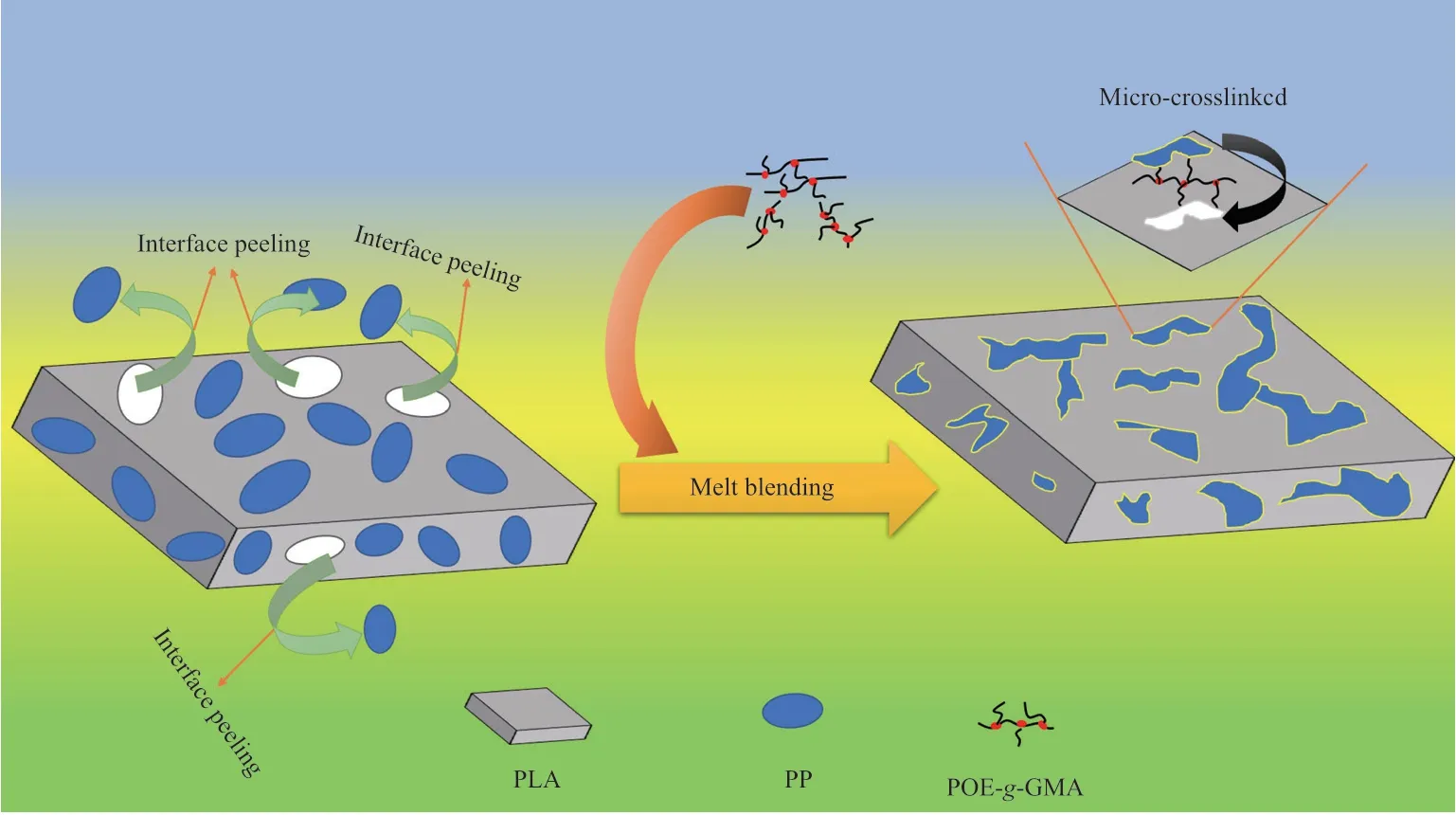
圖7 POE-g-GMA對PLA/PP界面調控機理示意圖Fig.7 Schematic diagram of the regulation mechanism of POE-g-GMA on the PLA/PP interface.
3 結論
1)POE-g-GMA的GMA基團與PLA的端羥基或羧基在共混過程中發生了化學反應。
2)POE-g-GMA含量超過2%(w)時,PLA/POE-g-GMA/PP在整個頻率范圍內均呈非牛頓特性。POE-g-GMA改善了PLA與PP的相互作用,使共混物的熔融行為得到了增強。
3)當POE-g-GMA的含量低于6%(w)時,主要起增塑作用,活化PLA分子鏈的運動能力,提高PLA的結晶度;當POE-g-GMA含量高于6%(w)時,反而降低了PLA的結晶能力,誘導共混體系向連續相結構轉變。
4)當POE-g-GMA添加量為6%(w)時,PLA/POE-g-GMA/PP共混材料的拉伸強度達到29.2 MPa,且斷裂伸長率提高至25%,為PLA/PP的10倍,材料剛韌平衡性良好。
猜你喜歡
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
當代陜西(2020年13期)2020-08-24 08:22:02
中國外匯(2019年17期)2019-11-16 09:31:14
制造技術與機床(2017年5期)2018-01-19 02:49:17
金秋(2017年4期)2017-06-07 08:22:16
中國材料進展(2016年10期)2016-12-26 06:50:20
濰坊學院學報(2016年2期)2016-12-01 13:00:11
新聞傳播(2015年11期)2015-07-18 11:15:04
現代企業(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
