P450 酶介導的EGFR-TKIs 藥物與其他藥物的相互作用*
2022-02-18 09:04:48柳迎華趙麗艷付青姐
中國藥業 2022年2期
關鍵詞:劑量
柳迎華,趙麗艷,付青姐
(中國人民解放軍海軍第九七一醫院藥劑科,山東 青島 266071)
表皮生長因子受體酪氨酸激酶抑制劑(EGFR-TKIs)多用于治療晚期非小細胞肺癌(NSCLC),在體內主要經P450 酶代謝,能影響P450 酶的活性,從而干擾其他藥物的體內代謝。該藥為口服劑型,使腫瘤居家治療成為可能,但相當一部分居家用藥腫瘤患者在服用EGFR-TKIs 的同時會聯用其他藥物,不恰當的聯用會引起藥物與藥物間的相互作用(DDI),從而影響藥物療效,產生嚴重不良反應。為此,總結了EGFR-TKIs 的代謝途徑、相關酶抑制劑/ 誘導劑對EGFR-TKIs 的影響、EGFR-TKIs對P450酶活性的影響及對相關底物的干擾,為臨床合理聯合用藥提供參考。
1 EGFR-TKIs 藥物及P450 酶
EGFR-TKIs 主要作用于EGFR 蛋白酪氨酸激酶,與三磷酸腺苷競爭性結合酪氨酸激酶功能區,從而阻止EGFR 下游信號通路的激活。截至2021年5 月6 日,國家藥品監督管理局(NMPA)共批準了7個EGFR-TKIs藥物用于治療晚期NSCLC,包括第1 代的吉非替尼、鹽酸厄洛替尼和鹽酸埃克替尼,屬可逆性EGFR-TKI,廣泛應用于EGFR 敏感突變的晚期NSCLC 治療;第2 代的阿法替尼、達可替尼,為不可逆的EGFR-TKI;第3 代的甲磺酸奧希替尼、甲磺酸阿美替尼等,能不可逆地抑制EGFR,且對T790M突變陽性有較好的治療作用。
EGFR-TKIs在體內主要經肝P450酶代謝。肝內常見代謝酶包括CYP1A2,CYP2A6,CYP2C9,CYP2C19,CYP2D6,CYP2E1,CYP3A4[1]。研究表明,50%以上的藥物經CYP3A4代謝,約25%的藥物經CYP2D6代謝,15%的藥物經CYP2C9 及CYP2C19 代謝[1]。P450 酶的活性受遺傳因素或藥物因素的影響,從而影響底物藥物的代謝。在藥物的代謝性相互作用中,由酶的抑制作用而引起的占全部藥物相互作用的70%,由酶的誘導而引起的占23%[2]。EGFR-TKIs 與P450酶抑制劑合用可增加EGFR-TKIs 的血藥濃度,加大不良反應的發生可能,嚴重者可導致治療中斷或延遲;而與P450酶誘導劑合用可增加EGFR-TKIs 的代謝,導致藥效下降,造成疾病進展或復發。此外,EGFR-TKIs 也可對P450 酶產生誘導或抑制作用,影響其他酶底物的代謝。故了解P450 酶在EGFR-TKIs 代謝過程中的作用對于臨床合理用藥尤為重要。
2 P450 酶介導的EGFR-TKIs 與其他藥物的DDI
2.1 吉非替尼
吉非替尼為首個應用于臨床的EGFR-TKIs,于2015年被NMPA 批準單藥用于具有EGFR 基因敏感突變的局部晚期或轉移性NSCLC 的治療。與傳統含鉑化學治療(簡稱化療)相比,吉非替尼可顯著延長中位無進展生存期(PFS),但不能顯著延長平均總生存時間(OS),主要應用于EGFR 突變陽性的晚期NSCLC 的一線治療。
吉非替尼在體內主要經CYP3A4 快速、大量代謝,少量經CYP2D6 及CYP3A5 代謝[3]。人血漿中共檢測到8 種吉非替尼代謝產物,主要產物O-去甲基-吉非替尼含量較少,且僅有較弱的抗腫瘤活性。吉非替尼原形及代謝產物主要經膽汁排泄[3]。
CYP3A4 強抑制劑可明顯抑制吉非替尼的代謝及清除,吉非替尼與CYP3A4 強抑制劑伊曲康唑聯用時,峰濃度(Cmax)增加51%,藥時曲線下面積(AUC)增加78%[4]。目前尚無相關專家推薦吉非替尼與CYP3A4 強抑制劑聯用時需調整吉非替尼的劑量。藥品說明書推薦聯用,但應嚴密監測吉非替尼的藥品不良反應。
CYP3A4 強誘導劑可明顯降低吉非替尼的暴露量,且這一改變具有臨床意義。吉非替尼與CYP3A4強誘導劑利福平合用時,Cmax降低65%,AUC降低83%[5]。吉非替尼與CYP3A4 中效誘導劑苯妥英合用時,Cmax降低26%,AUC降低47%[5]。臨床中含中藥成分如人參的藥物使用較多,應注意避免吉非替尼與其聯用[6]。藥品說明書及文獻資料均推薦,與CYP3A4中、強效誘導劑(如利福平、苯妥英、卡馬西平、圣約翰草等)合用時,若未出現嚴重不良反應,吉非替尼劑量可增至500 mg/d。終止CYP3A4 誘導劑7 d 后,重新按原劑量(250 mg/ d)給藥[7]。
CYP2D6 參與O-去甲基-吉非替尼的生成,在CYP2D6 慢代謝人群中,吉非替尼的平均暴露量增加了2 倍,但研究者認為該變化可能不具有臨床意義[8]。目前,使用吉非替尼前不建議進行CYP2D6 基因型檢查,CYP2D6 弱表達患者也無須調整吉非替尼劑量。但考慮CYP2D6 抑制劑可能增加吉非替尼的血藥濃度,建議聯用CYP2D6強抑制劑時,密切監測吉非替尼的藥品不良反應[7]。
體外研究表明,吉非替尼有弱酶誘導作用,可有限抑制CYP2D6(43%)及CYP2C19(24%)[9]。SWAISLAND等[4]的試驗中,吉非替尼與CYP2D6 底物(美托洛爾)合用,美托洛爾的暴露量增加35%,但不認為這一改變具有臨床相關性[4]。吉非替尼與其他治療窗較窄的CYP2D6 底物聯用時,仍建議密切監測CYP2D6 底物相關不良反應,必要時調整劑量[7]。
2.2 厄洛替尼
厄洛替尼與吉非替尼具有相似的臨床功效與應用,于2006年被NMPA批準單藥用于EGFR具有敏感突變的局部晚期或轉移性NSCLC 的治療,包括一線治療、維持治療或既往接受過至少1 次化療進展后的二線及以上治療。
厄洛替尼主要經肝內CYP3A4/ 5 代謝,肝外途徑(包括腸內的CYP3A4、肺內的CYP1A1、腫瘤組織的CYP1B1)對厄洛替尼的代謝也有一定作用[10-12]。其主要代謝產物為O-去甲基-厄洛替尼,具有與厄洛替尼相似的抗腫瘤活性。厄洛替尼原形及代謝產物主要經膽汁排泄。
CYP3A4 強抑制劑可顯著增加厄洛替尼的暴露量。厄洛替尼與CYP3A4 強抑制劑酮康唑聯用時,Cmax增加69%,AUC增加67%;與CYP3A4 和CYP1A2 的共同抑制劑環丙沙星合用時,Cmax增加17%,AUC增加39%,活性代謝產物的AUC增加60%(數據來自OSI制藥公司2014年發布的厄洛替尼處方信息)。目前還未明確該暴露增加的臨床相關性。提示厄洛替尼應盡量避免與CYP3A4強抑制劑、CYP3A4/ 1A2 共同抑制劑、強效CYP1A2 抑制劑(如氟伏沙明)合用,若必須合用,應嚴密監測不良反應,一旦發現毒性反應,建議以50 mg 的梯度進行減量[7]。
CYP3A4 強誘導劑可明顯降低厄洛替尼的暴露量。厄洛替尼與CYP3A4強誘導劑利福平合用時,AUC減少58%(數據來自OSI制藥公司2014年發布的厄洛替尼處方信息)。故應盡量避免厄洛替尼與CYP3A4 強誘導劑合用,若必須聯用,厄洛替尼可在密切關注不良反應的前提下加量至300 mg/d,若能良好耐受2 周以上,可考慮進一步增量至450 mg/d,并密切監測藥品不良反應。當停用相關誘導劑時,厄洛替尼劑量減少至初始量[7]。CYP3A4 的其他誘導劑如苯妥英、卡馬西平、巴比妥、圣約翰草等,也應盡量避免與厄洛替尼合用,如必須合用,應考慮調整厄洛替尼的用藥劑量。
吸煙可誘導CYP1A1/ 2 的活性[13],而厄洛替尼部分經CYP1A1/ 2 代謝,故吸煙也可導致厄洛替尼血藥濃度變化[14]。在實體瘤患者中,厄洛替尼的血漿清除率較已戒煙者及從未吸煙者高24%[15]。健康志愿者中,厄洛替尼的AUC增加2.8 倍,Cmax增加1.5 倍,24 h血漿濃度增加9 倍[14]。已證實正吸煙NSCLC 患者的厄洛替尼最大耐受量為300 mg,但與標準劑量(150 mg)相比,300 mg 劑量在化療失敗后的二線治療中未顯示療效提高。建議服用厄洛替尼的患者在用藥前就開始戒煙。
厄洛替尼為CYP1A1 的強抑制劑及CYP3A4/ 2C8的中效抑制劑。在CYP1A1 弱表達患者中,厄洛替尼的影響尚未明確。厄洛替尼與典型的CYP3A4 底物(如咪達唑侖、紅霉素)及CYP3A4/2C8底物(如紫杉醇[16])合用時,相關藥物的清除率未受影響,故認為與其他CYP3A4 底物清除間的相互作用也不可能發生。但厄洛替尼與CYP3A4底物華法林合用時,有大出血及國際標準化比值(INR)升高可能[17];與辛伐他汀合用時可增加辛伐他汀濃度,誘發橫紋肌溶解癥,與苯妥英合用加重其藥品不良反應[18-19]。因此,對于治療窗較窄的CYP3A4 底物,厄洛替尼與之合用時仍需注意監測藥品不良反應。
2.3 埃克替尼
埃克替尼為我國自主研發,于2011年被NMPA 批準用于EGFR 突變陽性的局部晚期或轉移性NSCLC 的一線治療。
參與埃克替尼代謝的酶包括CYP3A4/5,CYP2C19,CYP1A2[20]等6 個途徑,共檢測到19 種Ⅰ相代謝產物。代謝產物與原形藥主要經膽汁排泄。
80%以上的埃克替尼經CYP3A4/ 5 代謝[20],埃克替尼與強效CYP3A4 抑制劑酮康唑合用時,AUC增加3.22 倍;而與強效CYP3A4 誘導劑利福平合用時,AUC降低0.55 倍[20]。目前尚無文獻建議埃克替尼與CYP3A強誘導劑或抑制劑合用時需調整埃克替尼的劑量,但應充分意識到合用的風險。
3.7 %~7.5%的埃克替尼經CYP1A2 代謝[20],此酶代謝的藥物底物不多,但吸煙能誘導CYP1A2 酶的活性,對埃克替尼的藥動學及藥效學參數產生影響,建議使用埃克替尼且吸煙的患者及時戒煙。
埃克替尼對CYP2C9 和CYP3A4 有明顯的抑制作用。據報道,埃克替尼與CYP3A4 底物西羅莫司合用會誘發間質性肺炎[21],與CYP2C9底物華法林合用時出血風險增加[22]。提示埃克替尼與治療窗較窄的酶底物合用時,應監測藥品不良反應。
2.4 阿法替尼
阿法替尼于2017年被NMPA 批準用于治療EGFR敏感突變的晚期NSCLC。其在體內的代謝非P450途徑,其主要代謝產物為蛋白質共價加合物。此外,阿法替尼也并非CYP 酶系的誘導劑或抑制劑,故不太可能與P450酶底物發生相互作用[23]。
2.5 達可替尼
達可替尼于2019年5 月被NMPA 批準用于EGFR 19 號外顯子缺失突變或EGFR 21 號外顯子L858R 置換突變的局部晚期或轉移性NSCLC 的一線治療。與第1 代EGFR-TKIs 相比,達可替尼延長了治療失敗的時間(TTF),升高了客觀緩解率(ORR),并有延長OS的趨勢。
達可替尼主要經肝臟代謝,包括氧化作用及谷胱甘肽結合反應。氧化作用主要由肝臟CYP2D6 完成,生成與達可替尼藥理作用相似的O-去甲基-達可替尼[15]。CYP3A4 參與其他次要氧化代謝物的生成[24],代謝產物及原形主要經膽汁排泄。
研究顯示,合用時暴露量會增加,但無臨床意義。在健康志愿者中與CYP2D6 強抑制劑帕羅西汀合用時,達可替尼的暴露量增加37%,達可替尼與其代謝產物總AUC增加約6%。該結果不認為具有臨床相關性[25]。CYP2D6 慢代謝型患者的代謝與其他人群相似或最多增加10%,也無臨床意義[25]。故臨床合用達可替尼與CYP2D6 強抑制劑時,暫不需調整達可替尼的劑量。
對于CYP2D6 誘導劑與達可替尼合用暫無文獻報道,建議盡量避免合用,以免降低療效。
達可替尼為CYP2D6 的強效抑制劑。達可替尼與CYP2D6 底物右美沙芬合用時,右美沙芬的Cmax增加9.7 倍,AUC增加9.6 倍,而達可替尼的濃度未發生改變[24]。建議CYP2D6 的底物(尤其是治療窗較窄的底物)與達可替尼合用時,應調整藥物的劑量或使用其他替代藥物。
2.6 奧希替尼
奧希替尼于2017年被NMPA批準用于既往EGFR-TKI治療進展、T790M 突變陽性的局部晚期或轉移性NSCLC 患者。奧希替尼作為二線治療藥物仍可顯著提高T790M 陽性患者PFS 及腦轉移患者無病進展率,目前各指南推薦用于晚期NSCLC患者的一線治療。
奧希替尼主要經CYP3A 代謝[26],還可能存在并未完全明確的其他代謝途徑。臨床前研究共檢測到AZ7550和AZ5104這兩種具有藥理活性的代謝產物。
肝藥酶抑制劑可導致奧希替尼的代謝減少,但認為這一改變可能無臨床意義。一項在晚期NSCLC 中展開的研究表明,與CYP3A4 的強抑制劑伊曲康唑合用時,奧希替尼的Cmax減少20%,AUC增加24%,代謝產物AZ5104 的暴露量無明顯改變[27-28]。提示CYP3A4 強抑制劑可能不會顯著影響奧希替尼的代謝。
肝藥酶的誘導劑可導致奧希替尼的暴露量明顯減少。晚期NSCLC 患者合用奧希替尼和CYP3A4 的強誘導劑利福平時,奧希替尼的AUC下降78%,AZ5104 的AUC下降82%、Cmax下降78%[27-28]。提示應避免同時使用奧希替尼與CYP3A4強誘導劑,阿期利康制藥有限公司發布的2019年版甲磺酸奧希替尼藥品說明書建議,若必須合用,奧希替尼的劑量可增至160 mg/d,停止服用CYP3A4強誘導劑3周后,恢復至原劑量80 mg/d。中效CYP3A4 誘導劑也可降低奧希替尼的暴露量[27],也應慎用,盡量避免合用;奧希替尼與中效及低效CYP3A4誘導劑合用時,暫無相關劑量的調整要求。
體外研究表明,奧希替尼是CYP3A4/ 5 的競爭性抑制劑。奧希替尼與CYP3A4 底物(辛伐他汀)合用時,辛伐他汀的Cmax增加23%,AUC增加9%[29],但這一改變無臨床意義。因此認為奧希替尼與CYP3A4的底物合用時,相互作用的臨床意義并不顯著[27]。
2.7 阿美替尼
阿美替尼為全球第2 個第3代EGFR-TKI,是我國自主研發的創新藥物,也是全球首個中位PFS 超過1年(二線用藥)的第3 代EGFR-TKI。于2020年3 月被NMPA 批準用于既往經EGFR-TKIs 治療時或治療后出現疾病進展,且經檢測確認存在EGFR T790M 突變陽性的局部晚期或轉移性NSCLC 患者。臨床研究表明,阿美替尼在疾病控制方面與奧希替尼相似,但對顱腦癥狀的控制效果略遜于奧希替尼。
阿美替尼目前僅有Ⅰ期臨床數據尚未開展大型的代謝性臨床研究。江蘇膏森藥業集團有限公司發布的2020年版阿美替尼藥品說明書提示,該藥物在血漿中主要以原形存在,部分由肝臟CYP3A 酶代謝。CYP3A4強抑制劑可導致阿美替尼AUC增加3.1~4.0 倍,應慎與大環內酯類抗菌藥物、伊曲康唑等強CYP3A4抑制劑合用,目前無相關合用劑量調整的建議,建議盡量避免合用,若必須合用,嚴密監測不良反應。CYP3A 強誘導劑則可導致阿美替尼AUC減少約90%,故阿美替尼治療期間應慎用利福平、卡馬西平、苯妥英鈉、圣約翰草等CYP3A強誘導劑。
3 常見EGFR-TKIs 比較
常見EGFR-TKIs 的代謝途徑及酶抑制劑/ 誘導劑對其的影響,EGFR-TKIs對P450酶活性的影響及藥物相互作用,常見P450 酶敏感底物及對其活性的影響見表1至表3。

表1 常見EGFR-TKIs的代謝途徑及酶抑制劑/誘導劑對其的影響Tab.1 Metabolic pathways of common EGFR-TKIs and effect of enzyme inhibitors/inducers on them
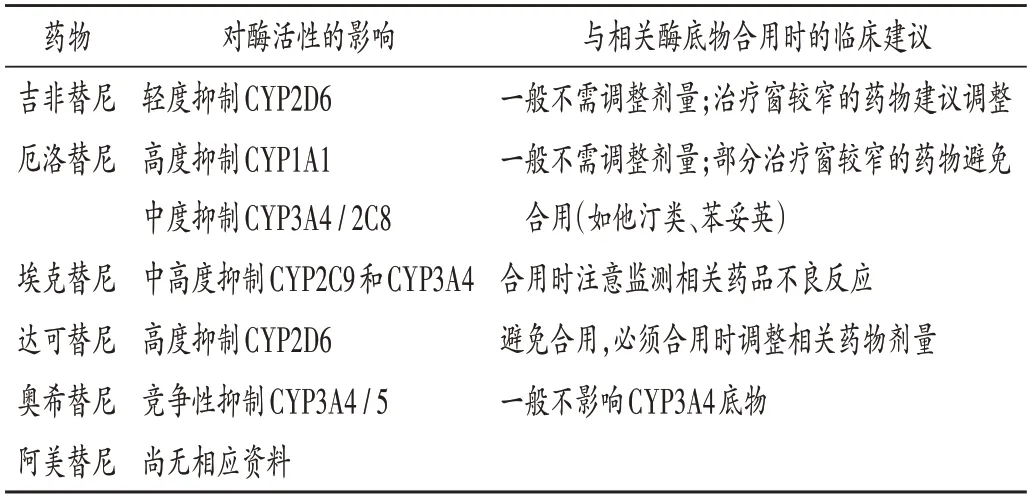
表2 EGFR-TKIs對P450酶活性的影響及藥物相互作用Tab.2 Effect of EGFR-TKIs on the activity of P450 enzyme and drug-drug interactions

表3 常見P450酶敏感底物及對其活性的影響Tab.3 Common P450 enzyme sensitive-substrates and the effect of different drugs on their activity
4 合理用藥建議
EGFR-TKIs能顯著延長NSCLC 患者PFS,OS等生存數據,從而成為EGFR 基因突變陽性患者的首選治療方案。但EGFR-TKIs 在體內主要經P450 酶代謝,并能對其產生抑制作用,這使得伴有一些基礎疾病的老年患者在長期合用其他藥物時會產生一系列的DDI。臨床常用的經CYP3A4代謝的他汀類、環孢素、卡馬西平等,經CYP2D6 代謝的美托洛爾、普萘洛爾、氟西汀等,在慢性病老年患者中使用率較高,且治療窗較窄,不適當合用的藥品不良反應發生率極高。
EGFR-TKIs主要經CYP3A,CYP2D6,CYP1A2代謝,當與酶誘導劑或抑制劑合用時,可能使藥效下降或不良反應加重,如厄洛替尼及埃克替尼均經CYP1A2 代謝,吸煙可誘導CYP1A2的活性,從而降低藥物的療效,故建議此類患者戒煙;西咪替丁為CYP3A4 及CYP2D6的抑制劑,大多數EGFR-TKIs 經此兩酶代謝,若合用可加重藥品不良反應。EGFR-TKIs 不僅經P450 酶代謝,同時也抑制了相關酶的活性,當一些敏感底物合用時,可使相應血藥濃度升高,從而導致嚴重藥品不良反應,如達可替尼可高度抑制CYP2D6 的活性,若聯用治療窗較窄的美托洛爾、右美沙芬、可待因、普羅帕酮合用時,可增加這些藥品不良反應。厄洛替尼可抑制CYP3A4的活性,部分他汀類調脂藥經CYP3A4代謝,若合用,建議選用不經CYP3A4代謝的瑞舒伐他汀。
綜上所述,臨床醫師在制訂肺癌靶向治療方案時,也應充分考慮患者是否同時合并其他基礎疾病,是否使用相關藥物,合理設計藥物方案,減少不必要的DDI,減少藥源性疾病的發生,提高藥物療效。臨床藥師應協助醫師提供藥物相互作用的藥物信息,做好用藥監護,共同保障患者的治療效果及用藥安全。
猜你喜歡
課堂內外·初中版(科學少年)(2023年10期)2023-12-10 00:43:06
全科護理(2022年10期)2022-12-26 21:19:15
中國合理用藥探索(2022年1期)2022-11-26 00:22:32
今日農業(2022年4期)2022-11-16 19:42:02
鄉村科技(2021年33期)2021-03-16 02:26:54
國際放射醫學核醫學雜志(2021年10期)2021-02-28 08:41:58
藥學與臨床研究(2015年4期)2015-06-05 11:35:54
衛生職業教育(2014年24期)2014-05-20 09:05:38
同位素(2014年2期)2014-04-16 04:57:20
中國合理用藥探索(2014年11期)2014-03-11 20:30:20
