1例德朗熱綜合征患兒的NIPBL基因突變分析
2022-01-26 13:02:50徐瑞張英懿劉心潔
山東醫藥 2022年2期
關鍵詞:數據庫
徐瑞,張英懿,劉心潔
1山東大學齊魯醫院兒科,濟南250012;2壽光市人民醫院兒科
德朗熱綜合征(CdLS)是一種以嚴重神經發育障礙為主要表現的遺傳綜合征,其在活產患兒中的發病率為1/10 000~1/30 000,主要為散發性,極少為家族聚集性發生。CdLS的臨床表現包括智力發育落后、特殊面容、宮內和生后生長遲緩及多臟器畸形等[1-3]。此外,有報道顯示,CdLS患兒體液免疫受到影響,較普通兒童更易罹患感染性疾病[4]。目前臨床上CdLS經典的致病基因有5種:NIPBL、SMC1A、SMC3、RAD21和HDAC8[5],隨著基因檢測的普及及檢測水平的提高,有更多的致病基因被發現。2020年8月,我們臨床確診1例CdLS患兒,為明確病因,應用高通量捕獲測序進行致病基因變異檢測,并用Sanger測序驗證測序結果以及致病基因的家系分析。現報告如下。
1 資料與方法
1.1 病例介紹 患兒女,2個月又23 d,因“發現頭圍偏小1 d”就診于我院門診。患兒系第1胎第1產,孕38+6周經陰道分娩,出生過程順利,出生體質量3 400 g,生后無窒息缺氧病史。出生后聽性腦干反應未通過,出生第42天復查通過。可追視距離眼前20 cm左右的黑白卡。無相關家族史。體格檢查:體質量4 150 g,頭圍33.7 cm(<-3 SD),前囟僅容指尖。多毛,眉毛濃密、長,后發際線低,耳廓邊緣及背部毛發多。長人中,短鼻,口角下歪,上唇薄,小下頜,高腭弓。胸廓無畸形,心肺無異常。軀干有兩處2 cm×1 cm咖啡斑,腳趾細長,無通貫掌。四肢肌力、肌張力可,生理反射正常存在,雙側巴氏征陰性。其母孕期第16周行唐氏篩查無異常,孕后期多次彩色超聲示胎兒宮內發育遲緩,雙頂徑<2 SD,家屬拒絕行羊水穿刺行基因檢查。實驗室檢查:患兒近期血常規、胸片、肝腎功能+心肌酶+生化、甲狀腺功能、血氨未見異常。血串聯質譜和尿氣相色譜—質譜聯用法未見異常。染色體微陣列(CNV)掃描分析未見異常(見OSID碼圖1)。
1.2 CdLS的判斷 根據2020年CdLS國際共識提出的臨床特征評分[6],分為基本特征(2分/項)和提示性特征(1分/項)。基本特征包括:①連眉和(或)濃眉;②短鼻、凹鼻嵴和(或)鼻孔前傾;③長人中和(或)人中扁平;④薄上唇紅和(或)嘴角下彎;⑤少指(趾)畸形和(或)先天性無指;⑥先天性膈疝。提示性特征包括:①全面發育遲緩和(或)智力障礙;②胎兒宮內發育遲緩(<-2 SD);③出生后生長遲緩(<-2 SD);④小頭畸形[產前和(或)產后];⑤小手和(或)短足;⑥第5指發育不全或不發育;⑦多毛。臨床特征評分≥11分,且包括至少3個基本特征者診斷為典型CdLS;臨床特征評分9~10分,且包括至少2個基本特征者診斷為非典型CdLS;臨床特征評分4~8分,且包括至少1個基本特征者,需結合分子測試結果進行診斷;臨床特征評分<4分者不診斷CdLS。
1.3 患兒及其父母全基因組檢測 經患兒監護人知情同意并簽署知情同意書后,抽取患兒及其父母外周血各2 mL,EDTA抗凝,使用QIAamp全血DNA提取試劑盒(德國Qiagen公司)提取基因組DNA。應用GenCap液相捕獲目標基因技術(北京邁基諾公司),捕獲與CdLS相關的包含CdLS相關基因(NIPBL、SMC3、RAD21、SMC1A和HDAC8)的編碼外顯子區域和側翼區域。利用Illumina Nextseq 500第二代測序儀捕獲到的區域進行雙端測序,讀長為150 bp。目標區域測序后,去除測序數據中的接頭和低質量數據。運用BWA軟件比對到參考基因組上(hg19版本),對測序深度、均一性、探針特異性等數據進行統計分析。
使用GATK軟件對該樣本的比對數據進行多態性位點檢測,對單核苷酸多態性(SNPs)和插入缺失突變(InDels)等數據進行統計和分析,查找SNPs及InDels在千人基因組數據庫、人類基因突變數據庫、Clinvar數據庫、ESP6500外顯子測序計劃數據庫、ExAC數據庫(ALL和EAS)及邁基諾內部1 000例正常漢族人群數據庫頻率,利用SIFT、PolyPhen2、MutationTaster、GERP+等數據庫對SNPs及InDels的致病性進行預測分析,篩選上述數據庫中頻率<0.05且預測結果均為致病性的位點作為與疾病相關的位點。
根據需要測序的DNA片段合成引物,用聚合酶鏈反應(PCR)法進行擴增,用ABI3730xl測序儀(美國Applied Biosystems公司)以Sanger測序法進行測序,將測序結果與目標區域捕獲測序后的結果進行比對。
2 結果
2.1 患兒CdLS臨床特征評分結果 患兒CdLS臨床特征評分11分,且包括4個基本特征,符合典型CdLS的診斷標準。
2.2 患兒及其父母全基因組檢測結果 全外顯子基因組測序檢出患兒NIPBL基因第6外顯子存在c.461G>A雜合錯義突變。NIPBL基因c.461G>A變異在各基因數據庫及近期文獻中均無該位點的相關報道,該位點變異為未報道過的新變異。生物信息學蛋白功能綜合性預測軟件REVEL預測結果為潛在有害,SIFT、MutationTaster、GERP+預測結果為有害、有害、有害。
經Sanger測序驗證,該變異存在于患兒及患兒父親,c.461G>A可導致編碼蛋白第154位精氨酸替代為谷氨酰胺(p.R154Q),見圖1。經家系驗證分析,其父該位點為雜合突變,其母該位點無變異,其父手臂處多毛,無特殊面容、智力發育落后、生長發育落后、多臟器畸形等表現。
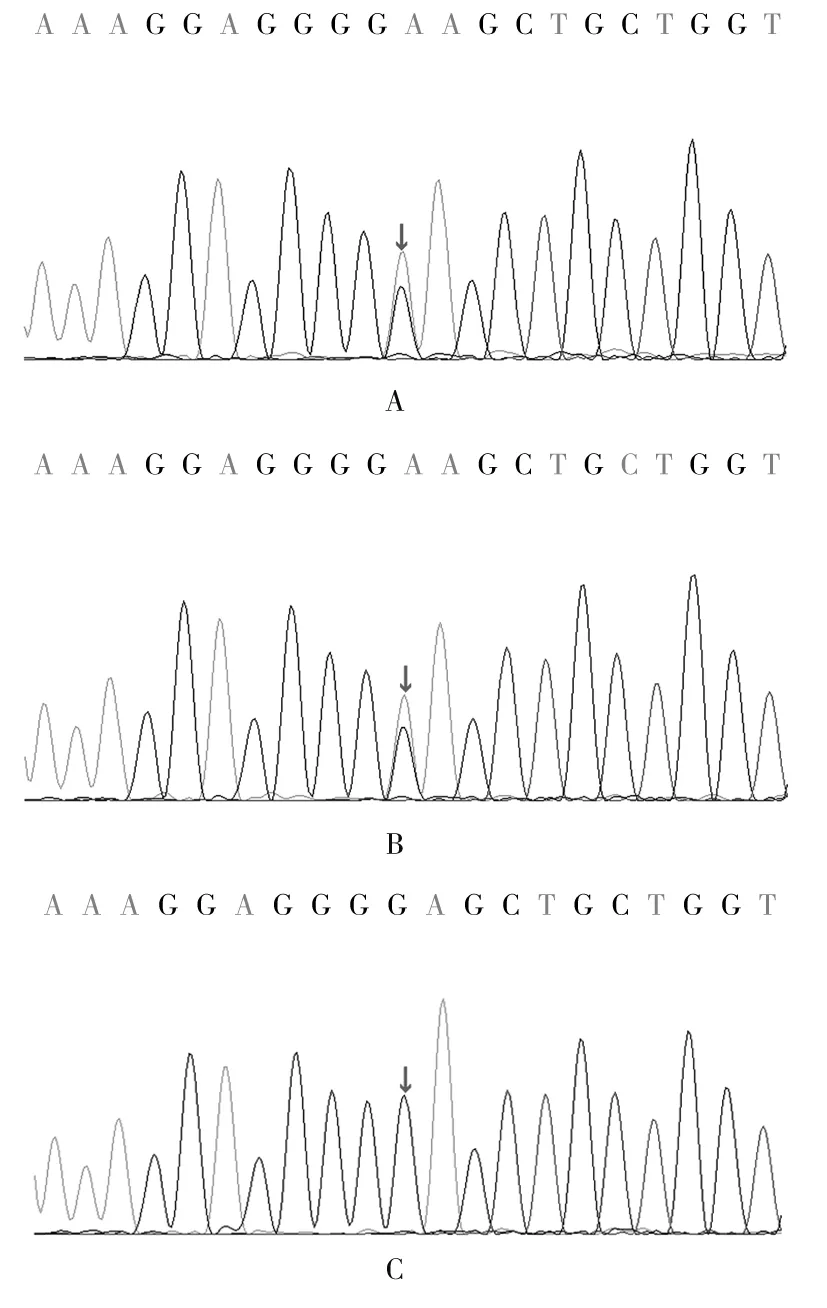
注:A為患兒,箭頭所指位置有A與G兩種核糖核苷酸,存在雜合變異;B為患兒父親,箭頭所指位置有A與G兩種核糖核苷酸,存在雜合變異;C為患兒母親,箭頭所指位置有G一種核糖核苷酸,無變異。
3 討論
CdLS是一種以嚴重神經發育障礙為主要表現的遺傳綜合征,多數為散發病例,<1%的患者有同樣受累的父母[6]。目前其診斷主要根據2020年CdLS國際共識提出的臨床特征評分,本例患兒宮內發育遲緩(1分),具有典型的特殊面容,如小頭畸形(1分)、前發際線低、濃眉(2分)、長睫毛、鼻梁凹陷(2分)、寬鼻尖、長且扁平的人中(2分)、薄上唇紅、嘴角下彎(2分)、多毛癥(1分),總評分共11分,且包括4個基本特征,可確診為典型的CdLS。
患兒因生后查體發現頭圍偏小就診于我院門診,經門診再次詳細查體評估后臨床診斷為典型CdLS,為進一步驗證臨床診斷,故行相關基因檢測。全外顯子基因組測序結果顯示,該患兒NIPBL基因第6外顯子存在c.461G>A雜合錯義突變;其父該位點為雜合突變,其父體征上除手臂出多毛外無特殊面容、生長發育落后表現;其母該位點無變異,為野生型。為驗證該突變位點與患兒病癥的關系,經生物信息學蛋白功能綜合性預測軟件REVEL分析,預測結果為潛在有害,SIFT、MutationTaster、GERP+預測結果均為有害。因此推測,NIPBL基因c461G>A(p.R154Q)錯義變異可能是該患兒的發病原因,因該變異在千人基因組數據庫、人類基因突變數據庫和Clinvar數據庫及近期文獻中未有該位點的相關報道,為未報道過的新變異,該新變異的檢出豐富了NIPBL基因變異譜。經Sanger測序驗證,該位點突變存在于患兒及患兒父親。該突變位點位于5號染色體上NIPBL基因中,遺傳方式為常染色體顯性遺傳,該患兒發病;攜帶同一基因的父親有部分陽性體征,但臨床特征評分<4分者不能診斷CdLS,推測患兒父親為外顯不全。常染色體顯性遺傳的家系中偶見攜帶致病基因突變個體不患病,表現為不完全外顯,該個體稱為頓挫型,頓挫型可不表現顯性形狀或部分表現,但可以把顯性基因傳遞下去,使后代具有顯性癥狀。在系譜中,由于頓挫型的存在常染色體顯性遺傳可以出現隔代遺傳。如BOYLE等[7]2017年報道了1例CdLS患者的母親及姨媽攜帶與患者相同的RAD21基因單堿基對缺失,但不符合CdLS的臨床診斷標準,考慮與RAD21基因相關的外顯不全有關。
CdLS是一種罕見的多系統遺傳疾病,目前臨床認可的與CdLS發病相關變異基因有NIPBL、SMC1A、SMC3、RAD21、HDAC8、BRD4、ANKRD11、AFF4、ARIDIB、EP300、KMT2A、NAA10、SETD5、SMARCB1、TAF6、ZMYND11、PHIP、MED13L、MAU2等[8-11]。不同的基因變異所導致的臨床表現存在一定的差異,其中70%的患兒發病與NIPBL基因變異有關,但并不是所有NIPBL基因變異都能導致CdLS。NIPBL編碼的Nipped-B-like(NIPBL)蛋白是Cohesin復合體的裝載蛋白,鑒定為真菌和蒼蠅姐妹染色單體內聚蛋白2(SCC2)的人類同源物,它與SCC2形成復合體,是黏著蛋白裝載到染色體上所必需的,在染色質結構和轉錄調控中發揮重要作用[12-13]。在其他的發病相關變異基因中,如SMC1A、SMC3,已被證實與黏著蛋白有直接關系,而如ANKRD11等編碼基因,則不直接相關。根據有無黏著蛋白相關基因突變,可將患兒分為經典型及非經典型,部分患兒可同時有經典型及非經典型相關基因突變。本例患兒基因突變位點位于NIPBL基因,考慮為經典型。
CdLS患兒除特殊面容、肢體畸形外,還可出現嚴重的智力障礙,包括交流能力下降、重復行為和自殘行為以及多動,未來可能發展為自閉癥。其中交流能力下降主要表現為口語的落后,多通過手勢來表達[14-15]。因此,早期確診并進行干預對提高患兒的語言行為能力、保持其心理健康具有重要意義。隨著產前檢查技術水平的提升,針對CdLS的產前診斷越來越多,胎兒宮內發育遲緩、特殊面容、多系統畸形(如心臟畸形、消化道畸形等)在產前胎兒超聲中均可發現。對發現以上臨床特征的胎兒,以及有CdLS家族史的高危人群,可對孕婦進行羊水穿刺、收集胎兒脫落的細胞行基因檢測以明確診斷,實現優生優育。
目前針對CdLS尚無有效的治療方法,主要采取對癥支持治療及康復訓練。根據系統受累程度不同,針對患兒的隨訪、管理是不同的,發育遲緩的患兒應及早進行康復治療干預,多臟器畸形如先天性心臟病等可進行針對性治療及護理。CdLS患兒應根據病情定期隨訪,隨訪項目根據患兒年齡、疾病嚴重程度及累及系統制定。該例患兒在門診康復治療1個月后發育評估報告較前好轉,但落后于正常同齡兒。研究顯示,CdLS的發病機制與多種信號通路相關,其中包括經典的WNT通路。近期有研究顯示,氯化鋰可通過活化經典WNT通路,對CdLS模型有改善作用,提出鋰作為CdLS的可能治療策略[16],為臨床治療提供了一種新的選擇。
綜上所述,CdLS臨床主要表現為智力發育落后、特殊面容、宮內和生后生長遲緩及多臟器畸形等,可結合基因結果進一步明確診斷。本例CdLS診斷明確,NIPBL基因c461G>A(p.R154Q)錯義變異可能是其發病原因,該變異為新發基因突變,豐富了NIPBL基因變異譜。
猜你喜歡
財經(2017年15期)2017-07-03 22:40:49
財經(2017年2期)2017-03-10 14:35:35
華東師范大學學報(自然科學版)(2017年1期)2017-02-27 13:41:08
財經(2016年15期)2016-06-03 07:38:02
財經(2016年3期)2016-03-07 07:44:46
財經(2016年6期)2016-02-24 07:41:51
財經(2015年3期)2015-06-09 17:41:31
財經(2014年21期)2014-08-18 01:50:18
財經(2014年6期)2014-03-12 08:28:19
財經(2013年6期)2013-04-29 17:59:30
