超高效液相色譜-串聯質譜法檢測 蒸餾酒中的3種非法添加物質
2022-01-20 06:31:58王東江黎姝蔓
現代食品 2021年23期
◎ 楊 針,王東江,黎姝蔓,鄧 雄
(重慶市永川食品藥品檢驗所,重慶 402160)
我國白酒多以高粱、小麥等谷物為原料發酵后蒸餾儲藏所制,以蒸餾酒為主導[1]。大多數白酒中含有一定比例的醛類、甲醇和雜醇油等物質,這些物質易引發飲酒者的上頭、口干等不適反應。名優白酒往往通過改進釀造工藝提升酸類、酯類等香氣物質含量,降低醛類等有害物質含量,然而,一些不法商販違規在白酒中添加頭痛粉(主要成分為對乙酰氨基酚、水楊酸和咖啡因)以緩解劣質白酒引起的頭痛等不適癥狀,投機取巧,以次充好。頭痛粉作為藥品被添加入白酒中本就是非法添加成分[2],其藥品說明及病例報道也明確表示其與酒精混服對肝功能有損傷[3-4]。
目前,針對頭痛粉及其主要成分出現在白酒中的不良現象報道較多,其作為食品中的非法添加物檢測采用是高效液相色譜法和氣相色譜-串聯質譜法[5-7],但檢測方法存在靈敏度不高、分辨率不高、回收率不高、操作復雜、需不斷更換流動相及分析時間長等不足,相關方法無法滿足高靈敏度與較強抗干擾能力檢測微量或痕量成分的需求。因此本研究旨在建立蒸餾酒中對乙酰氨基酚、水楊酸、咖啡因的UPLC-MS/MS檢測方法,為蒸餾酒中非法添加頭痛粉的準確、快速判定檢測提供科學依據,為市場監管部門執法提供有力的技術支撐,從而達到保障人們飲食用藥安全的 目的。
1 材料與方法
1.1 材料與試劑
水楊酸標準對照品,壇墨質檢-標準物質中心,批號為1921804,含量為99.7%;對乙酰氨基酚標準對照品,壇墨質檢-標準物質中心,批號為1201801,含量為98.0%;咖啡因標準對照品,壇墨質檢-標準物質中心,批號為0981904,含量為98.0%;蒸餾酒酒樣,市售;甲酸(色譜純),山東西亞化學股份有限公司;甲醇(色譜純),Merck KGaA。
1.2 儀器與設備
Agilent 1290型高效液相色譜儀(包括二元泵,在線脫氣機,自動進樣器),Agilent 6420型三重四極桿質譜儀(包括電噴霧離子源,Masshunter數據工作站),美國Agilent公司;XP205型萬分之一電子天平,瑞士梅特勒-托利多儀器公司;JA5003N型千分之一電子天平,上海佑科儀器儀表有限公司;HH-8型水浴鍋,江蘇盛藍儀器制造有限公司;Mtegral5型超純水機,美國密理博公司。
1.3 方法
1.3.1 標準物質溶液的配制
標準儲備液:分別精密稱定對照品對乙酰氨基酚10.50 mg,咖啡因10.60 mg,水楊酸10.90 mg置于 25 mL容量瓶中,用甲醇溶解并定容至刻度,搖勻。分別得到對乙酰氨基酚儲備液(411.60 μg·mL-1)、咖啡因儲 備液(415.52 μg·mL-1)和水楊酸儲備液 (434.69 μg·mL-1),-18 ℃低溫避光保存。
混合標準中間液:分別精密移取對乙酰氨基酚、水楊酸和咖啡因儲備液各2.5 mL于同一20 mL容量瓶中,用超純水稀釋定容至刻度并搖勻,得到混混合標準中間液,乙酰氨基酚、水楊酸和咖啡因的濃度分別為51.450 μg·mL-1、51.940 μg·mL-1和54.336 μg·mL-1, 4 ℃避光保存。
混合標準工作液:分別精密移取對乙酰氨基酚、水楊酸和咖啡因對照品儲備液各0.1 mL于同一20 mL容量瓶中,用超純水稀釋定容至刻度并搖勻,得到混合標準工作液,乙酰氨基酚、水楊酸和咖啡因的濃度分別為2.058 μg·mL-1、2.078 μg·mL-1和2.173 μg·mL-1,4 ℃避光保存。
混合標準曲線系列工作液:分別吸取適量體積的混合 標準工作液,用水稀釋,配制成濃度分別為0 ng·mL-1、 10 ng·mL-1、20 ng·mL-1、50 ng·mL-1、80 ng·mL-1、 100 ng·mL-1和120 ng·mL-1的系列標準工作溶液。
1.3.2 樣品前處理優化
準確稱取5 g試樣(精確至0.01 g)置于50 mL燒杯中,于60 ℃水浴加熱30 min,殘渣全部轉移至50 mL容量瓶中,用純水定容至刻度并搖勻,經0.22 μm水相微孔濾膜過濾,供液相色譜-串聯質譜分析。
1.3.3 色譜條件優化
色譜柱:ZORBAX RRHD SB-C18(2.1 mm×100 mm, 1.8 μm);流速:0.3 mL·min-1;柱溫:25 ℃;進樣量:2.0 μL。流動相及梯度洗脫程序,如表1所示。
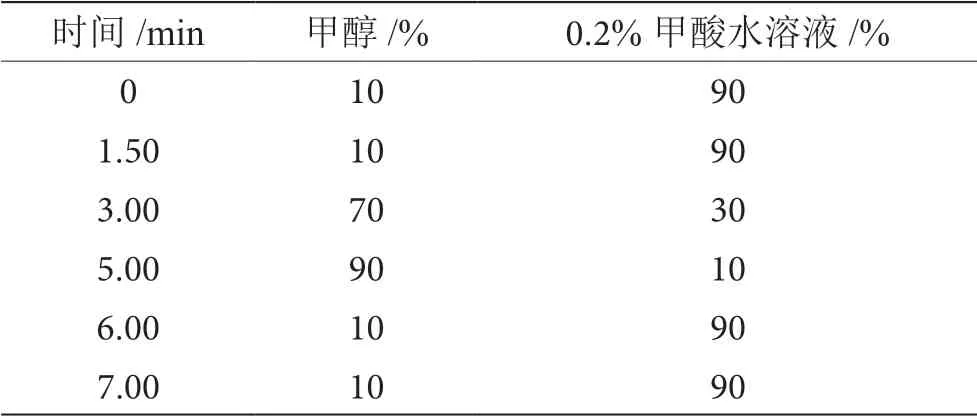
表1 流動相梯度洗脫程序表
1.3.4 質譜條件
電噴霧電離正負離子模式:ESI(±);掃描方式:多反應監測(MRM);離子化溫度:350 ℃;干燥器流量:10 L·min-1;噴霧電壓(IS):+4000 V/-3500V; 霧化器壓力:30 psi;監測離子及對應檢測參數如表2所示。
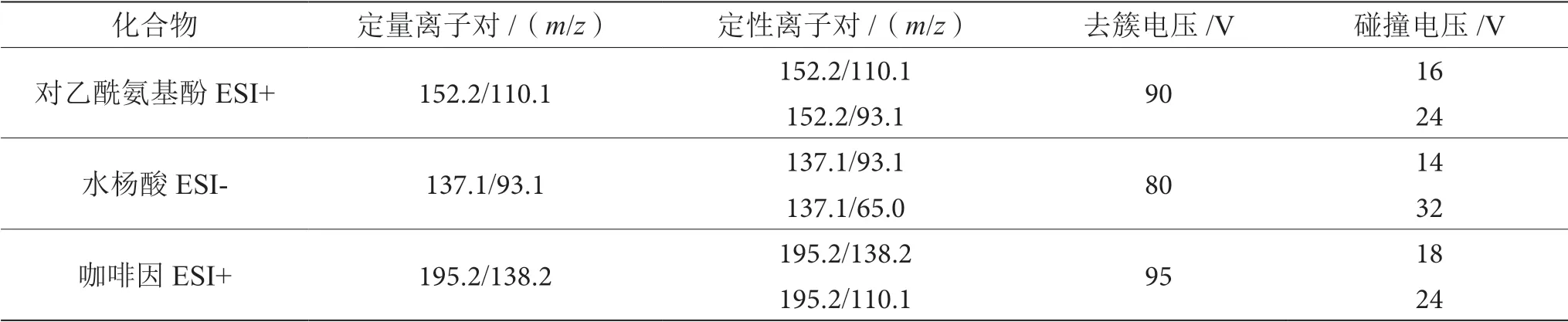
表2 監測離子及對應參數表
(1)定性測定。在相同的實驗條件下測定試樣溶液,若試樣溶液質量色譜圖中各物質的保留時間與標準溶液一致(變化范圍在±2.5%以內),且試樣定性離子的相對豐度與濃度相當的標準溶液中定性離子的相對豐度,其偏差不超過表3中規定,則可判定樣品中存在該物質。

表3 定性離子相對豐度的最大允許偏差表
(2)定量測定。將試樣溶液注入液相色譜-質譜/質譜儀中,得到各物質的定量離子峰面積,根據標準曲線計算試樣溶液中各物質的濃度。
2 結果與分析
2.1 檢出限與定量限的確定
將各組分標準物質逐級稀釋,找到信噪比S/N=3時各組分濃度,結果確定對乙酰氨基酚、水楊酸、咖啡因的檢出限分別為6 ng·mL-1、6 ng·mL-1、6 ng·mL-1,定 量 限(S/N=10)分 別 為20 ng·mL-1、20 ng·mL-1、 20 ng·mL-1。
2.2 標準曲線及相關系數
將配制好的標準系列溶液按照濃度由低到高的順序進樣測定,以各組分定量離子的色譜峰面積為縱坐標,以對應的濃度作為橫坐標,得到標準曲線回歸方程見圖1。
由圖1可得,對乙酰氨基酚標準曲線回歸方程為y=180.486176x+529.528216,相關系數為0.99936;咖啡因標準曲線回歸方程為y=53.704895x+50.217223,相關系數為0.99993;水楊酸標準曲線回歸方程為y=11.006995x+21.250089,相關系數為0.99989。
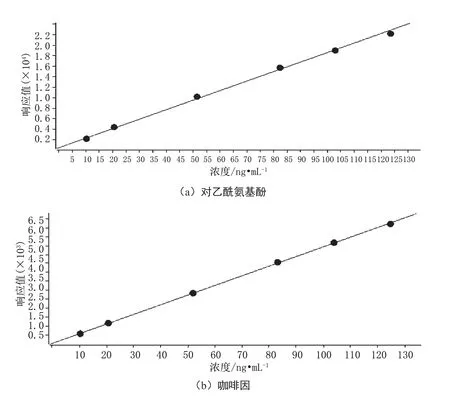
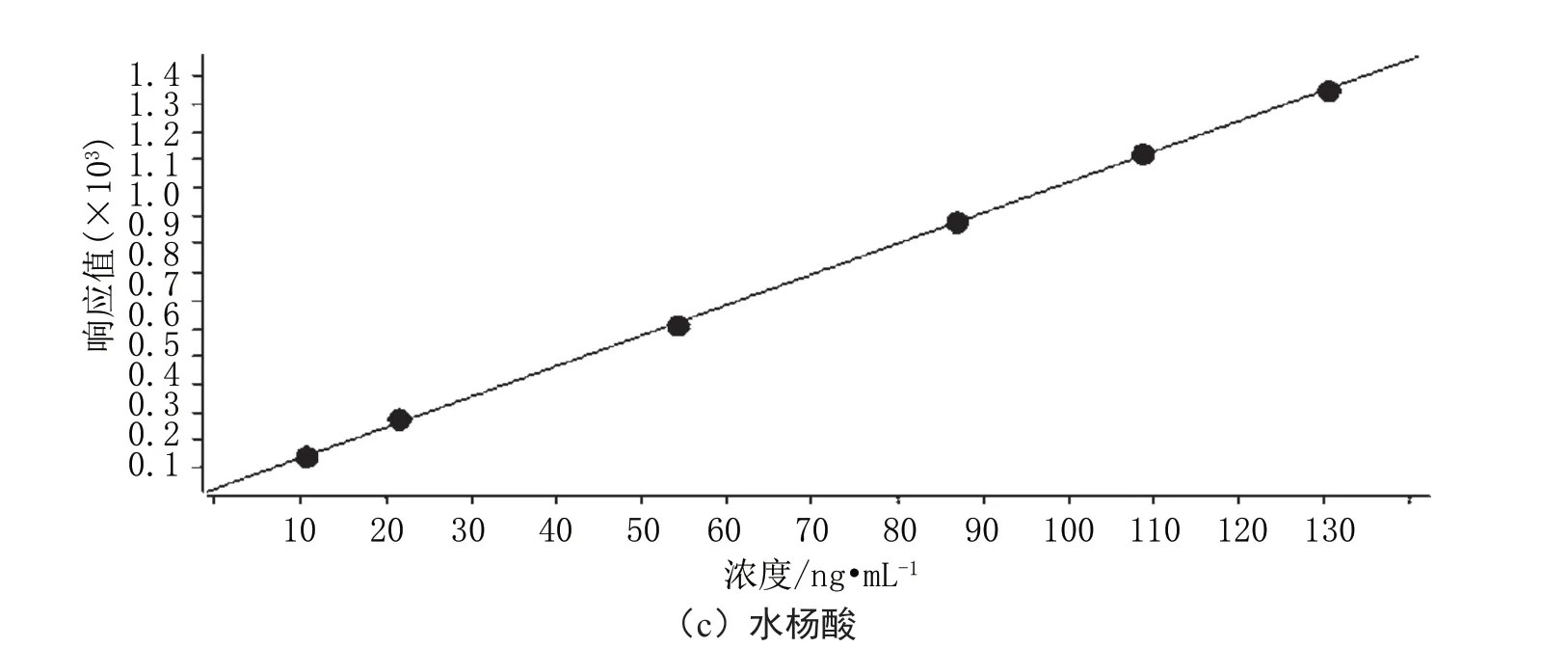
圖1 標準曲線回歸方程圖
2.3 檢測精密度及回收率
選用的蒸餾酒酒樣基質均為空白基質,本底值均為未檢出。
2.3.1 精密度
由表5可知,3種非法添加物質在空白基質中按照1倍、2倍、5倍定量限進行添加,樣品經處理后上機檢測,實驗結果精密度均≤3.0%,表明該方法處理的樣品含量結果精密度高。
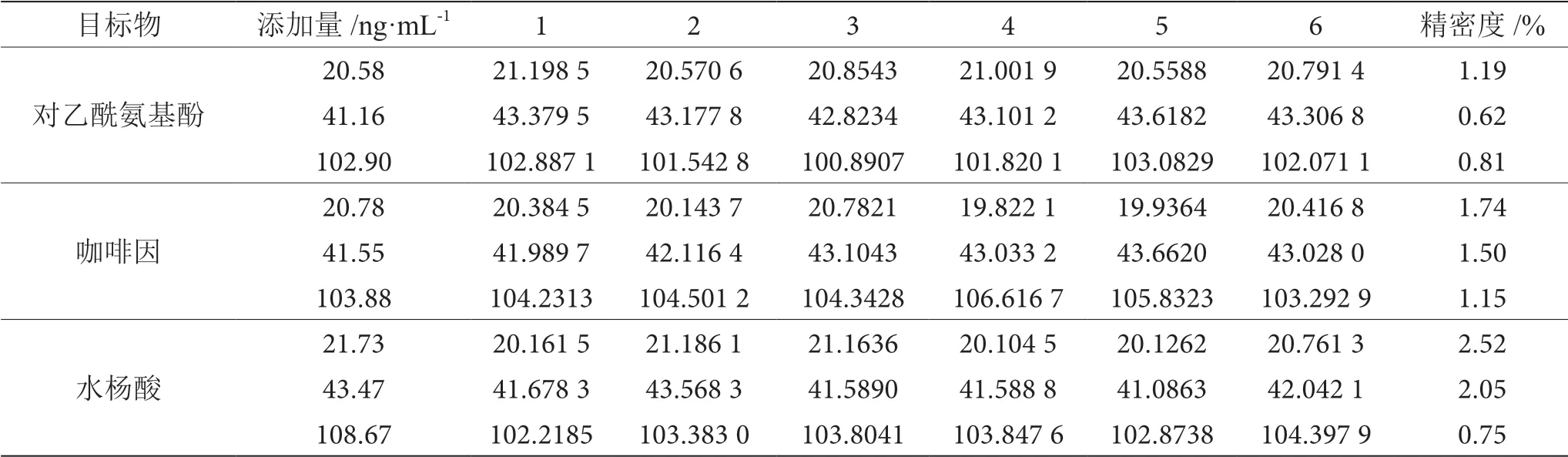
表5 對乙酰氨基酚、咖啡因、水楊酸檢測結果表
2.3.2 回收率
由表6可知,3種物質在不同的加標濃度水平下的平均回收率為95.2%~105.0%。

表6 對乙酰氨基酚、咖啡因、水楊酸回收率表
2.4 譜圖
50 ng·mL-1對乙酰氨基酚、水楊酸、咖啡因標準溶液的總離子流色譜圖見圖2。
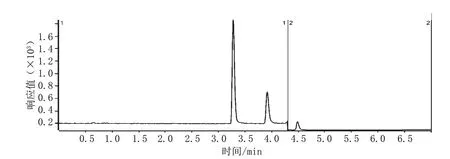
圖2 50 ng·mL-1對乙酰氨基酚、水楊酸、咖啡因標準溶液的總離子流色譜圖
50 ng·mL-1對乙酰氨基酚、水楊酸、咖啡因標準溶液的提取離子色譜圖見圖3。
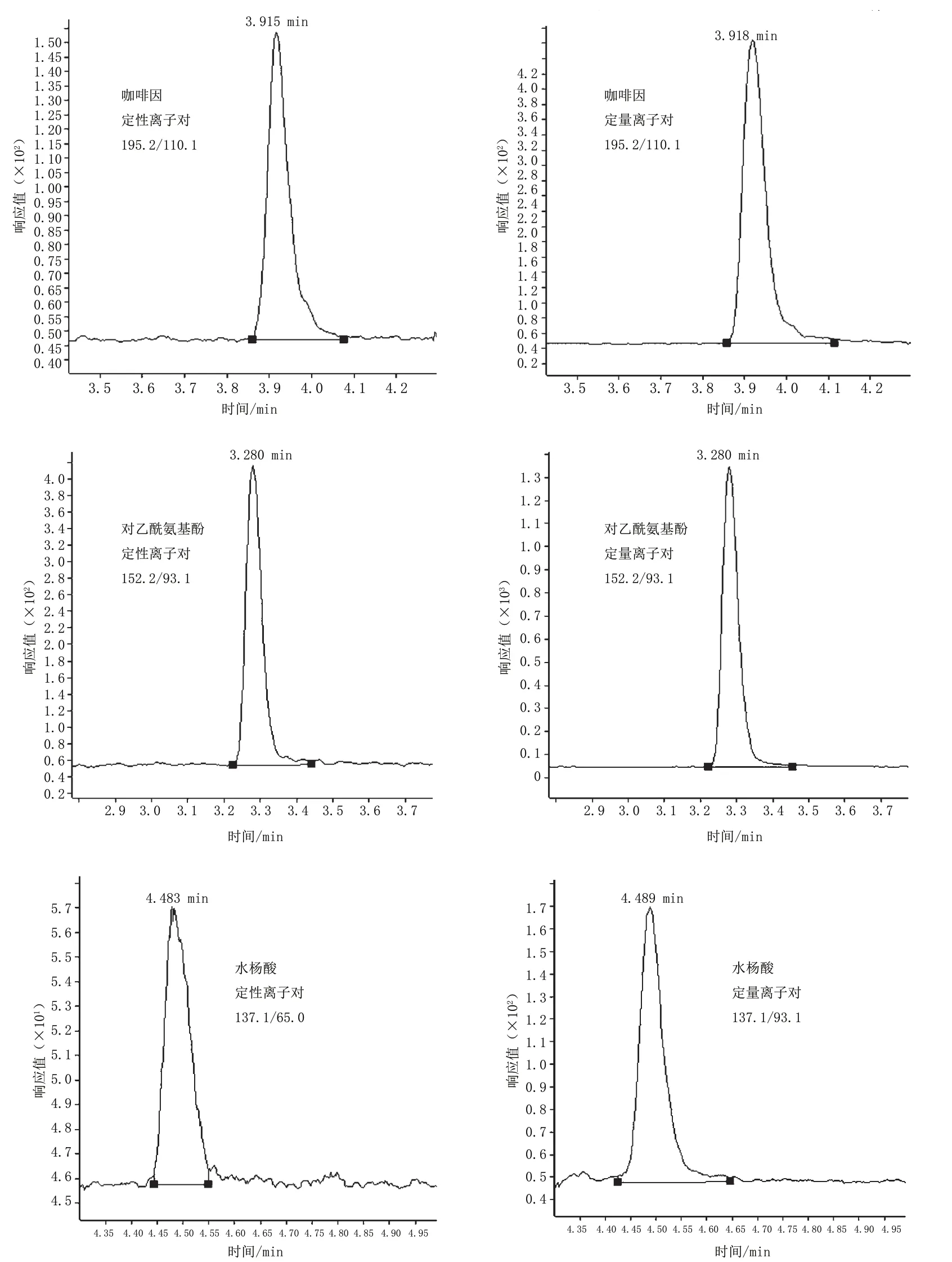
圖3 50 ng·mL-1對乙酰氨基酚、水楊酸、咖啡因標準溶液的提取離子色譜圖
2.5 樣品測定結果
采用本檢驗方法對市售的20種白酒進行了檢測,均未檢出非法添加對乙酰氨基酚、水楊酸、咖啡因。
3 結論
蒸餾酒中非法添加物質的查處和檢測能夠完善市場監管需求。本文用超高效液相色譜-三重四極桿串聯質譜法檢測蒸餾酒中頭痛粉的3種主要成分(包含水解成分),即對乙酰氨基酚、水楊酸、咖啡因。結果顯示,該方法靈敏度、加標回收率高,重復性好,不僅對蒸餾酒中非法添加對乙酰氨基酚、水楊酸、咖啡因的分析檢測方法起到了優化作用,還為食品安全的市場監管工作提供了準確有效的科學依據。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
當代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
海峽科技與產業(2016年3期)2016-05-17 04:32:12
