遠端型遺傳性運動神經病BSCL2基因突變1例
2022-01-13 09:27:24韓笑峰劉沛霖馬文彬王雪貞
濱州醫(yī)學院學報 2021年6期
關鍵詞:基因突變
黎 敏 韓笑峰 劉沛霖 馬文彬 王雪貞
濱州醫(yī)學院附屬醫(yī)院神經內科 山東 濱州 256003
遠端型遺傳性運動神經病(dHMN)是一組高度遺傳異質性疾病,由Nelson和Amick等于1966年首次報道,目前尚無其發(fā)病率的準確統(tǒng)計,且國內外報道較少。1993年Harding提出了一種基于遺傳模式和表型對dHMN進行分類的標準,共有7個亞型,其中遠端型遺傳性運動神經病V型(dHMN-V)呈常染色體顯性遺傳,由BSCL2或GARS突變所致,主要累及上肢[1]。本文報道1例與dHMN相關的BSCL2基因新突變(Exon7,c.742G>A,p.Val248Ile),并結合病例特點對相關文獻進行復習,以期提高臨床醫(yī)師對該病的認識水平,為dHMN的早期診斷提供病例參考。
1 病例資料
患者男,49歲。因“四肢肌肉萎縮、肌束顫動伴言語不清1年半”于2018年11月入院。2017年5月無明顯誘因出現(xiàn)雙上肢肌肉萎縮、肌束顫動,以遠端為著(圖1A、1B),自覺雙上肢無力,活動及持物可。就診于本院門診,針電極肌電圖提示所測肌肉右側三角肌、肱二頭肌及拇短展肌均可見正銳波、纖顫及二聯(lián)、三聯(lián)及多聯(lián)電位,輕收縮運動單位時限及大力收縮均未見明顯異常。給予營養(yǎng)神經治療后患者癥狀無緩解。2018年4月開始出現(xiàn)雙下肢(遠端為著)及肩胛區(qū)肌肉萎縮、肌束顫動(圖1C、1D),癥狀始終輕于上肢。2018年7月,出現(xiàn)舌肌輕度萎縮及震顫,伴言語不清,講話鼻音。上述癥狀逐漸加重。患者否認家族史。入院體檢:神志清楚,對答切題,言語不清,構音障礙,鼻音,雙側瞳孔等大等圓,對光反應靈敏,眼球各向活動自如,無凝視及眼震,雙側面紋對稱,伸舌居中,可見舌肌輕度萎縮及震顫,雙上肢、雙下肢及肩胛區(qū)可見肌束顫動,雙手骨間肌、大小魚際肌、雙側三角肌及肩胛區(qū)肌群、雙側脛前肌肌肉萎縮,四肢肌力5級,肌張力正常,雙下肢腱反射減弱,感覺及共濟檢查未見明顯異常,雙側巴氏征未引出。輔助檢查:血、尿、便常規(guī),甲功三項,睪酮,免疫九項,ANCA,抗核提取物抗體未見明顯異常。腦脊液檢查未見明顯異常。頭顱MRI及DWI提示腦內多發(fā)缺血灶。頸椎MRI提示C2/3、C6/7椎間盤突出。基因檢測顯示患者BSCL2基因異常,為c.742G>A(編碼區(qū)第742號核苷酸由G變?yōu)锳)的雜合核苷酸變異,位于Exon7,該變異導致了編碼蛋白的第248號氨基酸由Val變成了Ile(p.Val248Ile),為錯義突變(圖2A)。患者的長女23歲,攜帶與患者相同的基因突變(圖2B),尚未發(fā)病。其幼女14歲,未檢測到特定的突變(圖2C)。患者入院后予以改善微循環(huán)、營養(yǎng)神經、抗炎及對癥支持治療,癥狀無明顯好轉。出院后患者雙側肢體萎縮、肌束顫動及言語不清呈漸進性加重,逐漸出現(xiàn)負重及雙手精細活動不能,2020年5月復查EMG示右側正中、尺神經運動傳導未引出肯定波形,右側正中、尺神經感覺傳導均在正常范圍,雙側脛前肌、右側拇短展肌、左側脊旁肌T10、T11、胸鎖乳突肌均可見自發(fā)電位,MUP時限增寬,其中右側拇短展肌募集無力,雙側脛前肌募集力弱,提示廣泛神經源性損害。診斷:dHMN。
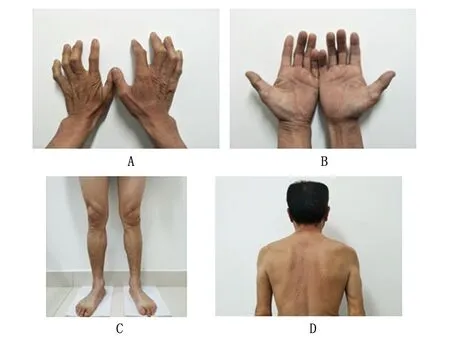
A. 雙手骨間肌明顯萎縮;B. 雙手大小魚際肌明顯萎縮;C. 雙下肢肌肉萎縮;D.肩胛下肌明顯萎縮。
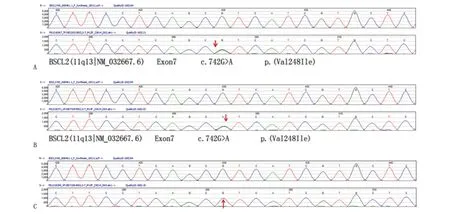
患者(A)及其長女(B)基因測序結果顯示c.742G>A(p.Val248Ile)雜合突變(箭頭所示);患者幼女(C)基因測序未檢測到特定的突變(箭頭所示)。
2 討論
dHMN又稱遠端型脊肌萎縮癥(dSMA),臨床罕見,主要表現(xiàn)為肢體遠端肌肉緩慢進行性無力、萎縮,而無明顯的感覺異常,多呈常染色體顯性遺傳,也可呈常染色體隱性遺傳或X連鎖隱性遺傳,亦可散發(fā)。近年來隨著全基因組測序技術的不斷進步,已鑒定出與dHMN相關的至少11個致病基因和4個基因組,但仍有超過80%的dHMN患者尚未發(fā)現(xiàn)明確的基因突變[2]。dHMN亞型之間的高度臨床異質性很大程度上源于不同的基因突變。
dHMN-V主要表現(xiàn)為上肢受累,常自一側或雙側上肢起病,手部肌肉如大魚際肌,小魚際肌,手背骨間肌等肌肉萎縮無力及遇冷攣縮,累及或不累及下肢遠端,查體可見腱反射減弱或消失,肌電圖檢查提示神經源性損害,本病具有遺傳異質性,進展緩慢,目前國內僅有少數(shù)幾個家系被報道[3-5]。研究發(fā)現(xiàn)BSCL2或GARS基因突變均可導致dHMN-V,已鑒定的BSCL2基因常染色體顯性遺傳突變有p.Asn88Ser,p.Ser90Leu,p.Ser90Trp和p.Arg90His[6-8]。
人類BSCL2基因位于染色體11q13,編碼一種稱為seipin的完整膜蛋白,定位于真核細胞內質網。seipin在睪丸及神經組織(大腦和脊髓)中高度表達[9]。目前,關于dHMN中BSCL2突變分子發(fā)病機制的大部分研究集中在由未折疊蛋白反應(UPR)激活引起的功能毒性增加[10]。突變的seipin由于其錯誤折疊引起內質網應激,啟動UPR,使細胞通過停止進一步的蛋白質翻譯和促進蛋白質重折疊或降解來降低內質網中未折疊蛋白質負荷。如果這些措施無法緩解內質網應激,UPR則啟動促凋亡途徑進而導致運動神經病變[2]。這可能是dHMN的發(fā)病機制。p.Asn88Ser和p.Ser90Leu突變位于3號外顯子(Exon3),發(fā)生在一些上肢發(fā)病的dHMN患者中。研究發(fā)現(xiàn),BSCL2中的p.Asn88Ser和p.Ser90Leu錯義突變破壞N端糖基化位點,導致錯誤折疊的蛋白質形成,表達這兩種突變蛋白的細胞上調BIP和HCOP(UPR的標志物),引起細胞凋亡增加[10],導致致命的神經退行性綜合征。
本例患者為中年男性,緩慢起病,臨床表現(xiàn)為四肢肌肉萎縮、肌束顫動,遠端為著,上肢重于下肢,伴言語不清,具有下運動神經元損害表現(xiàn)的體征,病情呈進行性加重,EMG檢查提示廣泛的神經源性損害,感覺系統(tǒng)始終未受累,基因檢測結果提示BSCL2錯義突變,綜合臨床表現(xiàn)及輔助檢查結果診斷為dHMN。
在dHMN患者中,由于BSCL2基因外顯率不完全,臨床表型及遺傳有廣泛的變異性,不同位點在致病機制和臨床表現(xiàn)上存在很大的差異,提示本病的基因型和臨床表型之間關聯(lián)復雜。關于BSCL2基因突變可引起dHMN早已得到證實,而本例患者的BSCL2基因p.Val248Ile突變,至今尚未見報道。患者長女具有相同的基因突變,目前尚未出現(xiàn)臨床癥狀,有待觀察和隨訪。此突變位點的致病性仍需更多的臨床案例印證,但其提示在已鑒定的突變之外存在尚未可知的致病突變。遺傳學檢測對dHMN診斷具有重要價值,有助于揭示其復雜的分子病理機制,這些研究的發(fā)展必將使得dHMN的預防與治療成為可能。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫(yī)學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現(xiàn)代檢驗醫(yī)學雜志(2016年4期)2016-11-15 02:01:14
中國現(xiàn)代醫(yī)學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫(yī)學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
