基于簡化基因組技術的啤酒花栽培種和野生種SNP位點開發及遺傳結構分析
2021-10-25 06:29:20趙亞琴樊叢照張際昭邱遠金辛海量李曉瑾張本剛王果平
中草藥 2021年20期
關鍵詞:新疆
趙亞琴,樊叢照,張際昭,邱遠金,辛海量,李曉瑾,張本剛,王果平*
1.新疆中藥民族藥研究所,新疆 烏魯木齊 830002
2.第二軍醫大學,上海 200433
3.中國醫學科學院藥用植物研究所,北京 100193
啤酒花Humulus lupulusL.隸屬桑科(Moraceae)葎草屬HumulusL.,是新疆特色的藥食同源植物,其體內含有的樹脂類、黃酮類、黃腐酚等化學成分具有抗病毒、抗氧化等藥用價值,同時啤酒花也是生產啤酒的基本原料,在保持啤酒風味及延長啤酒保質期等方面發揮著重要的經濟價值[1-2]。野生啤酒花主要分布在新疆阿勒泰地區的額爾齊斯河及其分支流域,如樺樹森林公園、哈巴河、布爾津、塔城地區和伊犁地區[3]。目前,在全國各地均有商業化種植。新疆不僅是啤酒花的原產地之一,也是我國啤酒花的主要產區,但據調查,目前新疆栽培種植的啤酒花主要是美國和日本引進的品種,在生產上存在品種單一、病蟲害嚴重等問題[2]。為培育本土品種,實現本地資源利用的最大化,探究新疆野生資源的遺傳多樣性及其與外來品種之間的遺傳差異性,有助于了解新疆本地野生資源的遺傳潛力,為其遺傳育種提供理論指導。目前對啤酒花遺傳特性的研究相對較少[5-6],特別是對其遺傳背景、新疆野生種與栽培種的關系等方面的研究,野生資源遺傳信息的缺乏也成為制約其遺傳育種的瓶頸。有研究表明,野生個體和栽培個體的遺傳多樣性在化學成分上存在明顯差異。所以探究新疆野生啤酒花植物的遺傳特征,明晰栽培個體與野生個體之間的遺傳背景及其遺傳關系,對培育地方品種、加強野生資源的利用具有重要意義。
隨著高通量測序技術的快速發展,基于第二代測序技術的簡化基因組測序specific-locus amplified fragment sequencing(SLAF-seq)、restriction-site associated DNA sequencing(RAD-seq)、genotyping by sequencing(GBS)等推動了進化生物學的又一步快速發展[7]。其中,SLAF-seq是一種高通量、高分辨率的SNPs位點識別與分型技術,是簡化基因組測序的一次革命[8-10]。SLAF-seq技術利用生物信息學方法,對目標物種的參考基因組進行系統分析,設計一個合適的酶切方案,構建SLAF-seq文庫,篩選出特異性長度片段,再應用高通量測序技術獲得高通量標簽序列,然后對數據分析,獲取滿足要求的SLAF片段。這些片段可以充分代表全基因組的序列特征信息,依據這些片段可以開發出大量的分子標記特別是單核苷酸多態(SNP)[11]。SLAF測序技術具高通量、高精度、短周期等優點,已經被運用于遺傳定位、高密度遺傳連鎖圖譜構建及不同個體間的多態性分析、系統進化和種質資源鑒定等領域[12-13]。本研究基于SLAF-seq測序技術以及獲取在新疆同域分布的20個啤酒花的野生種及18個栽培個體的大量多態性SLAF標簽,進而開發特異性強、穩定性高的的群體SNP位點。基于這些SNP位點從基因組水平明晰其野生種與栽培種之間的親緣關系及遺傳結構。旨在分析啤酒花不同個體間的遺傳分化,探討啤酒花野生種與栽培種之間的親緣關系,為其野生資源育種提供科學依據。
1 材料
本研究在野生啤酒花集中分布區采集20個野生個體,其中7份來源與阿勒泰地區,5份來源于塔城地區,1份來源于伊犁地區,3份來源于新源縣,4份來源于昌吉地區;18個栽培個體主要來源于阿勒泰地區、昌吉地區、哈密市及南疆的焉耆縣、沙雅縣及喀什地區。經中國醫學科學院藥用植物研究所張本剛研究員鑒定為啤酒花H.lupulusL.。采樣過程中選擇生長健壯、無病蟲的植株,采集其幼嫩的葉片并記錄經緯度,采集的新鮮樣品迅速用硅膠迅速干燥,存放于-80 ℃冰箱備用。
2 方法
2.1 基因組DNA制備
本實驗采用3×CTAB法提取分布在38個地點啤酒花的總DNA。采用1%瓊脂糖凝膠電泳進行電泳檢測DNA的產量和質量,確保所提取基因組DNA質量達到建庫要求,利用Thermo核酸檢測儀(Nanodrop 2000/2000c)測定DNA的濃度[14]。
2.2 酶切方案設計
根據已發布的啤酒花基因組信息(ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/831/365/GCA_000831365.1_hl_SW_version_1.0.fasta/GCA_000831 365.1_hl_SW_version_1.0.fasta_genomic.fna.gz).(基因組大小2.05 Gb,GC含量38.57%)作為參考基因組,組裝出的基因組大小為2.05 Gb,GC含量為38.57%。利用北京百邁客生物技術公司自主研發的酶切預測軟件對參考基因組進行酶切預測,最終選擇Rsa I和HaeIII酶對基因組DNA酶切,酶切片段長度在364~414 bp的序列定義為SLAF標簽,SLAF標簽在基因組上基本分布均勻。最終獲得酶切片段(SLAF標簽)數滿足預期標簽數[15]。
2.3 測序及產出數據的質量分析
基于位于重復序列的酶切片段比例盡可能低,酶切片段在基因組上盡量均勻分布,酶切片段長度與實驗體系吻合程度等原則設計酶切方案,對檢測合格的各個地點的啤酒花個體基因組DNA分別進行酶切。對得到的酶切片段(SLAF標簽)進行3’端加A處理、連接Dual-index測序接頭、PCR擴增、純化、混樣、切膠選取目的片段,文庫質檢合格后用Illumina測序平臺進行測序。為評估酶切實驗的準確性,選用水稻日本晴Oryza sativaL.ssp.japonica作為對照進行測序。對測序得到的原始數據進行識別、過濾、質檢、評估等分析,獲取各個個體的序列(reads)。
2.4 SLAF標簽和SNP標記的開發
利用Dual-index[16]對測序得到的原始數據進行識別,得到不同個體的reads分離的等位基因。過濾測序reads的接頭后,進行測序質量和數據量的評估。通過Control數據評估RsaI和HaeIII酶的酶切效率,以此判斷實驗過程的準確性和有效性。本試驗測序產生reads來源于都是不同地點的啤酒花在同一限制性內切酶的作用下產生的長度相同或相近的酶切片段,根據各個序列的相似度將38個個體的reads進行聚類,聚類到一起的reads來源于同一個SLAF標簽[17-18]。同一SLAF標簽在不同個體間的序列相似度遠高于不同SLAF標簽間的相似度;在同一個SLAF標簽中存在不同個體間序列的差異(即有多態性),即可定義為多態性SLAF標簽。以每個SLAF標簽中深度最高的序列類型作為參考序列開發全基因組范圍的SNP標記,對開發出的SNP根據完整度>0.5,MAF>0.05的標準進行篩選,最后利用篩選出的具有代表性的高質量SNP進行遺傳進化樹分析[19]、遺傳結構分析[20]和主成分分析(principal component analysis,PCA)[21]。
利用北京百邁客生物技術公司開發的軟件計算遺傳多樣性,利用AMOVA分析種群內、中群建和居群間的分子差異[22]。
3 結果與分析
3.1 建庫評估
SLAF-seq測序reads為基因組DNA的酶切片段,其堿基分布會受到酶切位點和PCR擴增的影響,測序reads的前2個堿基會呈現與酶切位點一致的堿基分離,后續堿基分布會呈現不同程度的波動(圖1)。

圖1 啤酒花測序堿基分布Fig.1 Sequence distribution of H.lupulus
結果表明本研究雙端比對效率在97.71%,比對效率基本正常。酶切效率是評價簡化基因組實驗是否成功的一個關鍵指標。基因組上的復雜結構區域(如環狀結構域、連續酶切位點等)、基因組DNA樣品純度較低、酶切時間不足等因素都可能影響限制性內切酶的活性,導致部分酶切位點未被切開。通過統計測序reads插入片段中殘留酶切位點的比例,統計比例越高,酶切效率越好。本實驗中水稻日本晴數據的酶切效率為89.92%,雙端比對效率為97.71%,酶切效率為89.92%,表明酶切反應及SLAF建庫均為正常。
3.2 測序數據統計與評估
本研究采用讀長126 bp×2作為后續的數據評估和分析數據。測序質量值(Q)是評估高通量測序單堿基錯誤率的重要指標,測序質量值越高對應的堿基測序錯誤率越低。如果某堿基測序出錯的概率為0.001,則該堿基的質量值Q應該為30(Q30)。對38個地點個體的測序數據進行統計,包括reads數量、Q30和GC含量。測序平均Q30為93.42%,平均GC含量為42.53%。由于所測序列的Q30數據較高,表明堿基出錯率很低,測序結果可靠(表1)。
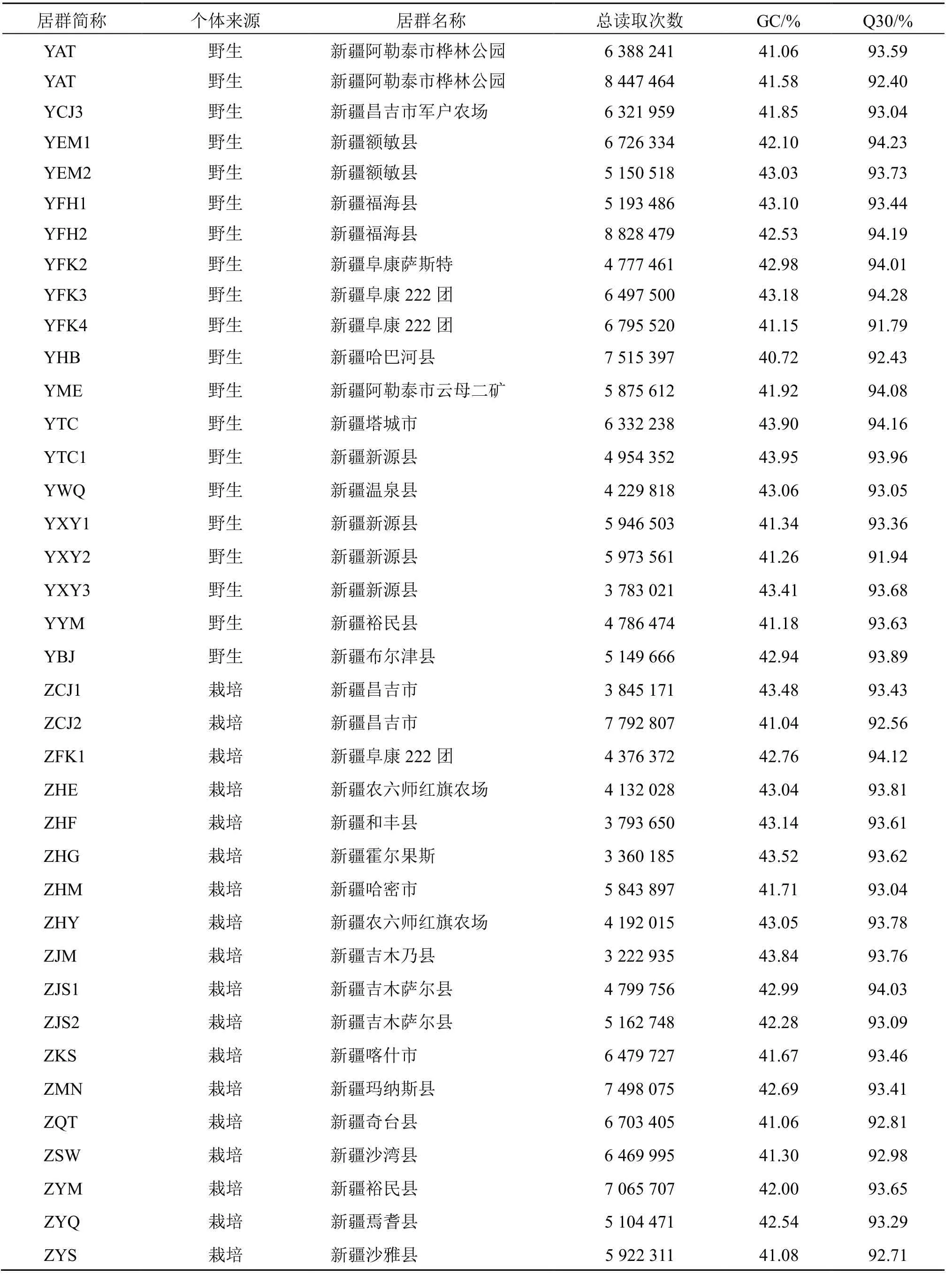
表1 啤酒花測序質量(Q30)及GC含量Table 1 Content of Q30 and GC of H.lupulus
3.3 SLAF標簽與SNP標記的鑒定
通過序列分析,從38個地點的啤酒花基因組中共獲得了863 228個SLAF標簽。標簽的平均測序深度為13.40 X,其中,多態性SLAF標簽有443 922個,共獲得2 867 140個高質量的群體SNP標記(表2)。
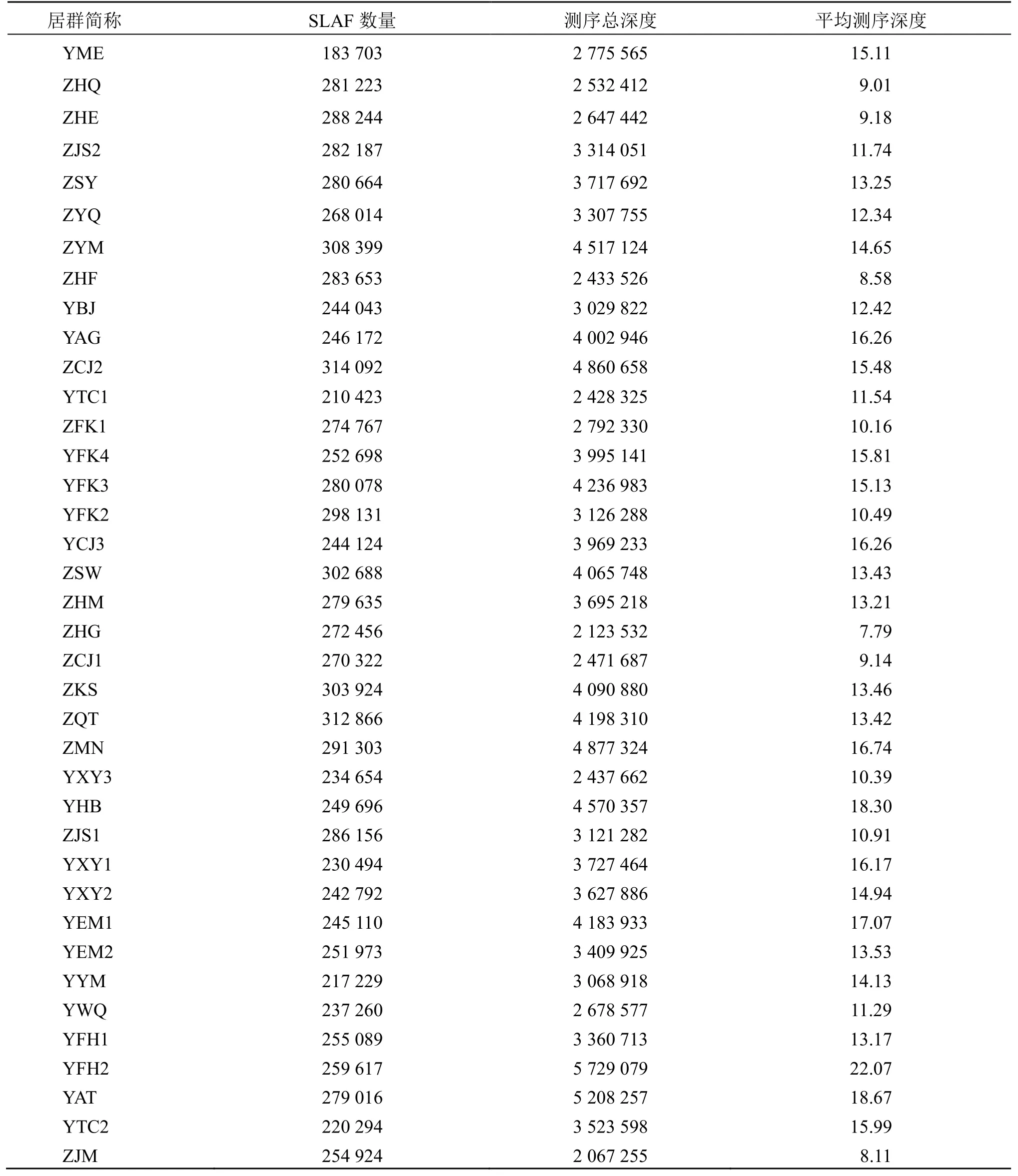
表2 啤酒花SLAF標簽Table 2 Label of SLAF of H.lupulus
3.4 系統發育分析
基于2 867 140個SNP位點對38個不同地點啤酒花的野生種與栽培種進行系統發育分析,結果表明38個啤酒花個體大致形成2個主要類群,類群I包括阿勒泰地區、塔城地區、伊犁地區的16個野生個體,類群II主要包括所有的栽培個體及阜康及昌吉地區的4個野生種(YFK2、YFK3、YCJ3、YFK4,圖2)。聚類分析顯示栽培個體與多數野生個體各自單獨聚成一類,表明栽培個體與野生個體之間的親緣關系較遠。

圖2 基于鄰接法的啤酒花的進化樹Fig.2 Evolutionary trees of H.lupulus based on NJ method
3.5 遺傳結構及PCA分析
基于開發出的2 867 140個SNP位點分析38個地區啤酒花的遺傳結構。交叉驗證聚類結果表明,當K=2時,交叉驗證錯誤率最低,可將38份啤酒花可劃分成2個不同的類群(圖3),結果與系統發育結果一致。S1類群(綠色)包括16個地區的野生個體,S2類群(黃色)包括18個地區的栽培個體及4個野生個體。結果表明野生個體與栽培個體之間存在較大的遺傳差異,形成了較明顯的遺傳分化。而在阜康及昌吉地區的栽培啤酒花品種來源多樣,遺傳組成比較復雜。

圖3 交叉各個K值對應的個體聚類圖及不同K值所對應的的交叉驗證錯誤率Fig.3 Admixture individual cluster values corresponding to each K value and admixture validation error rate corresponding to different K values
基于開發的SNP位點對20個野生個體及18個栽培個體進行PCA分析(PC1和PC2的累積方差貢獻率為35.89%),如圖4所示,20個野生個體和18個栽培個體在空間上表現出明顯的分離趨勢,說明野生個體與栽培個體之間具有明顯的遺傳差異性。其中野生個體分布在主成分坐標軸的右側,且彼此緊密地混聚在一起,說明采集的野生資源遺傳基礎總體較為狹窄,而栽培個體由于其品種來源多樣,遺傳背景復雜,表現出比野生個體更廣泛分布的特征,并且不同地區間的栽培個體之間也表現出較大的差異性。

圖4 PCA分析Fig.4 Analysis of principal components
3.6 遺傳多樣性與遺傳分化
38個啤酒花個體總的Shanon-Wiener指數為0.397,Nei多樣性指數為0.249。野生個體與栽培個體的遺傳多樣性具有一定的差異,整體表現為野生個體總的遺傳多樣性(0.454)大于栽培個體總的遺傳多樣性(0.398);總的Nei多樣性指數為0.249,其中野生個體的Nei多樣性指數(0.293)大于栽培個體(0.250)。分子方差分析表明野生個體與栽培個體之間存在較大的遺傳分化,其遺傳變異主要來源于野生個體與栽培個體之間。
4 討論
簡化基因組測序技術通過尋找合適的限制性內切酶來降低基因組的復雜程度,可顯著降低測序成本,同時還能獲得數量可觀的基因組變異信息并且能有效地克服基因組復雜的問題,已經被應用于多種植物的遺傳變異研究上[24-25]。基于SALF-seq簡化基因組數據對啤酒花的20個野生個體及18個栽培個體的遺傳結構分析表明,野生個體與栽培個體整體上存在較明顯的遺傳分化,但是在昌吉阜康區域的野生個體與栽培個體之間的遺傳分化并不顯著,存在一定的基因交流。這可能與當地進行大范圍的野生資源育種相關,尤其是昌吉地區啤酒花種植面積可達13.3 km2[26],大面積的栽培種植也增加了野生種之間的基因交流。
從聚類分析及PCA分析均表明,絕大多數栽培個體與野生個體(昌吉及阜康地區除外)之間存在明顯的遺傳差異,在DNA水平上單獨聚類,形成各自的遺傳結構。其中個別野生個體與當地栽培個體具有較近的親緣關系,說明不同類群間,尤其是利用野生資源育種的栽培個體和野生個體之間存在一定的基因交流。可見栽培種與野生種之間非絕對的生殖隔離,可以通過雜交的方式獲得變異植株,實現啤酒花種質創新和遺傳基礎拓寬。利用遺傳信息對新疆啤酒花的野生個體與栽培個體進行遺傳多樣性分析的報道較少,原俊鳳等[5-6]利用分子標記技術對新疆4個野生居群的遺傳多樣性研究結果表明新疆的野生啤酒花居群具有很高的遺傳多樣性。本研究也發現新疆野生啤酒花個體的遺傳多樣性高于栽培個體,豐富的遺傳資源是野生啤酒花育種中不可或缺的寶貴資源,同時也為新疆野生啤酒花資源的利用及本土品種的培育提供了一定的指導價值。
利益沖突所有作者均聲明不存在利益沖突
猜你喜歡
國畫家(2022年2期)2022-04-13 09:07:46
四川文學(2021年4期)2021-07-22 07:11:54
新作文·高中版(2021年4期)2021-07-14 11:55:06
環球人物(2021年8期)2021-05-06 08:00:43
音樂天地(音樂創作版)(2020年12期)2021-01-11 01:13:40
音樂天地(音樂創作版)(2020年7期)2020-09-11 06:17:18
華人時刊(2018年15期)2018-11-10 03:25:32
人民中國(日文版)(2015年10期)2015-04-16 03:53:48
絲綢之路(2014年9期)2015-01-22 04:24:46
新疆人文地理(2009年7期)2009-09-29 09:56:14
