熔融制樣-X射線熒光光譜法同時測定稀土礦石中螢石、重晶石及天青石
2021-10-11 13:03:36李可及張麗軍熊文良
理化檢驗-化學分冊 2021年9期
李可及,張麗軍,2,熊文良,2
(1.中國地質科學院 礦產綜合利用研究所,成都 610041;2.中國地質調查局稀土資源應用技術創新中心,成都 610041)
部分地方(如德昌、西昌等地)的稀土礦床會伴生螢石(主要成分為氟化鈣)、重晶石(主要成分為硫酸鋇)、天青石(主要成分為硫酸鍶)等非金屬礦[1],經選礦分離后,這些非金屬礦會賦存于尾礦中,如果能將其回收利用,則有助于提高礦山的效益,而分析測試是其相關工作的重要內容。
一般采用傳統方法測定螢石、重晶石、天青石含量:先單獨獲取一種非金屬礦,再通過一定手段排除干擾,如采用多次過濾分離操作排除硫酸鈣、碳酸鈣、碳酸鍶、碳酸鋇等的干擾,然后再通過乙二胺四乙酸(EDTA)容量法[2]、重量法[3]、火焰原子吸收光譜法(FAAS)[4-5]和X 射線熒光光譜法(XRFS)[6]等進行測定,由于前處理過程比較復雜,傳統方法的分析效率較低。本工作以傳統的分離方法為基礎[2-5],以含鈣和鍶的乙酸溶液處理樣品,僅需兩次過濾即可分離干擾,然后以熔融制樣-XRFS測定稀土礦石中氧化鈣、氧化鋇和氧化鍶的含量,并將其換算為氟化鈣、硫酸鋇和硫酸鍶的含量來表征螢石、重晶石和天青石等3種礦石的含量,以期為稀土礦石尾礦的回收利用提供參考。
1 試驗部分
1.1 儀器與試劑
Axios max 型X 射線熒光光譜儀,配功率為4 kW 的陶瓷薄鈹端窗(50μm)超尖銳銠靶X 射線管、Super Q 5.3A 軟件系統;TNRY-01C 型全自動熔樣機;鉑-金坩堝(wPt∶wAu=95∶5)。
重晶石礦石成分分析標準物質GBW 07812、GBW 07813;螢石成分分析標準物質GBW 07251、GBW 07253;水系沉積物標準物質GBW 07310;實驗室內部稀土礦石標準樣品REO-1、REO-2、REO-4。
混合熔劑:由質量比為12∶22的四硼酸鋰和偏硼酸鋰混合而成。
含鈣和鍶的乙酸溶液:稱取0.20 g 氯化鍶、2.00 g碳酸鈣于300 mL燒杯內,加入100 mL乙酸及適量水,加熱溶解,煮沸驅盡二氧化碳,冷卻后轉移至500 mL容量瓶中,以水定容,混勻備用。
二水合硫酸鈣、硫酸鍶、乙醇、乙酸為分析純;氟化鈣、碳酸鍶為光譜純;氯化鍶的純度為99%;碳酸鈣和二氧化硅的純度大于99.99%;試驗用水為去離子水。
以重晶石礦石成分分析標準物質(GBW 07812、GBW 07813)、硫酸鍶、碳酸鍶、螢石成分分析標準物質(GBW 07251、GBW 07253)、實驗室內部稀土礦石標準樣品(REO-1、REO-2、REO-4,可用稀土氧化物替代)及水系沉積物標準物質GBW 07310以一定的濃度梯度混合制備校準用標準樣品系列,按照試驗方法熔融,每種目標氧化物均制備12個以上濃度水平的標準樣品,其質量分數見表1。
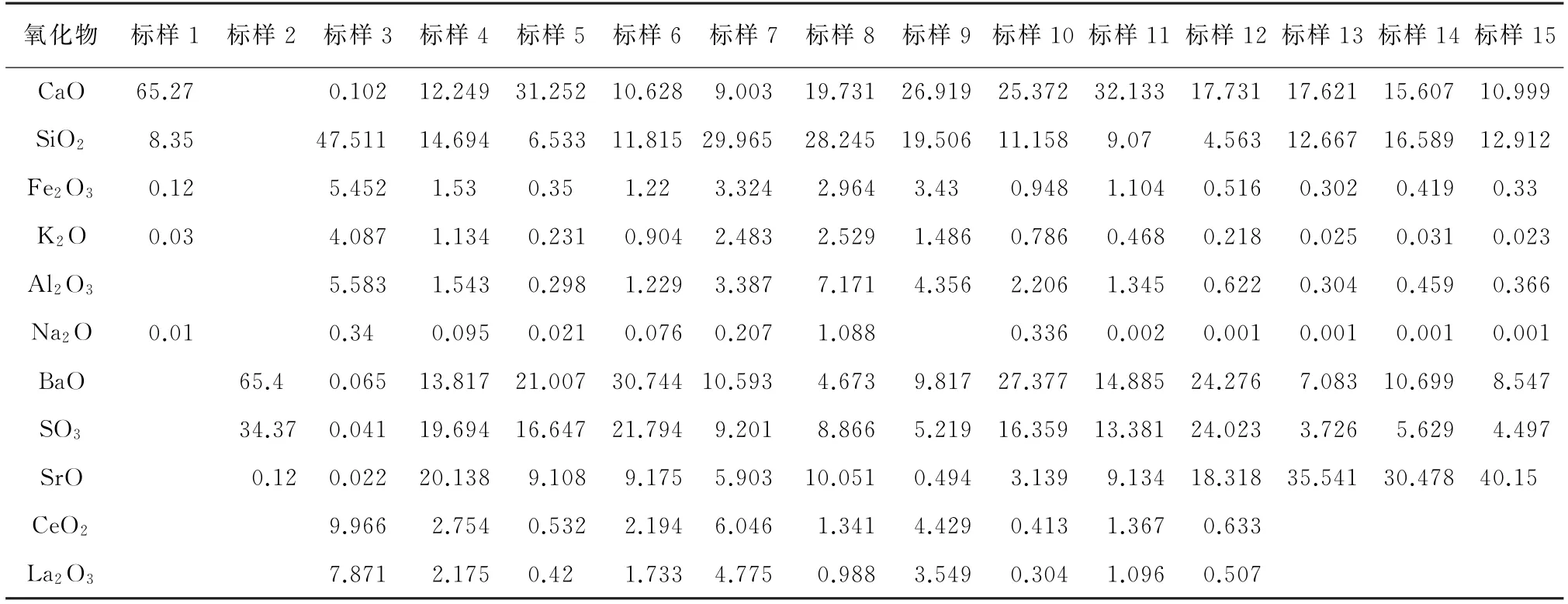
表1 標準樣品系列中11種氧化物的質量分數Tab.1 Mass fractions of 11 oxides in standard sample series %
1.2 儀器工作條件
XRFS的主要工作參數見表2,其中Flow 代表流氣正比探測器,Scint.代表閃爍探測器,鍶元素采用400μm 黃銅濾光片,其他元素均未采用濾光片。
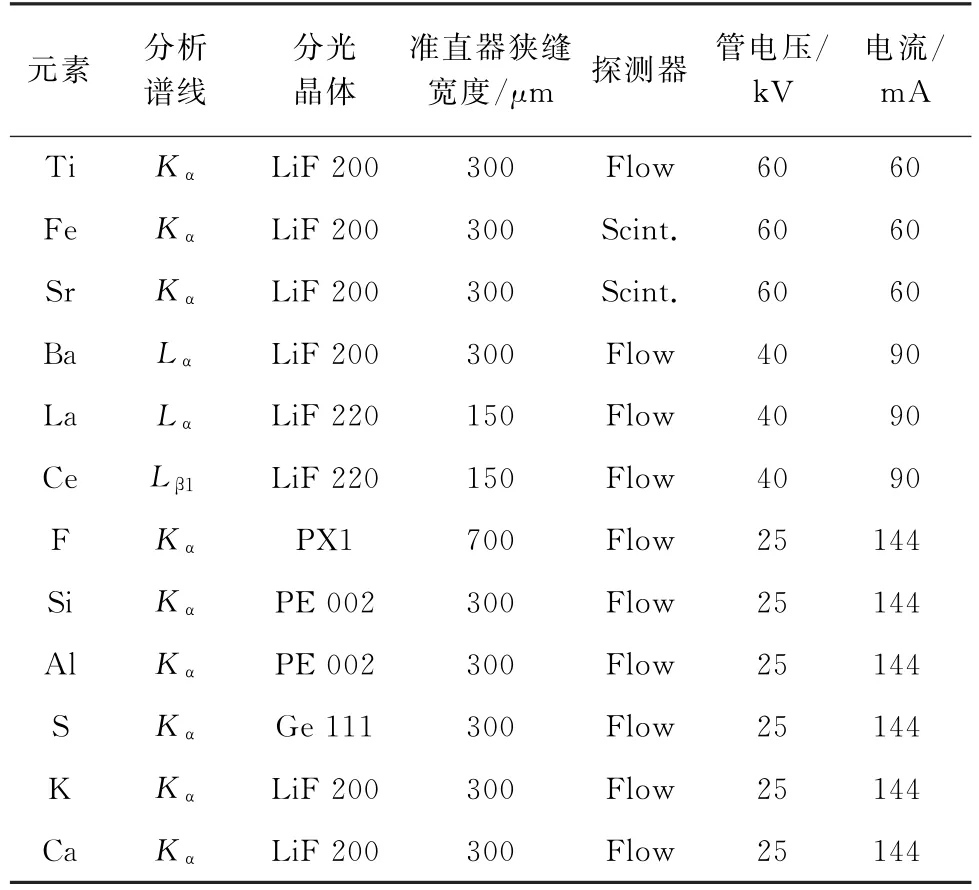
表2 XRFS工作參數Tab.2 Working parameters of XRFS
1.3 試驗方法
參考DZ/T 0130.2-2006?地質礦產實驗室測試質量管理規范 第二部 分巖石礦物分析試樣制備?制備樣品粉末。稱取0.800 0 g處理好的樣品于200 mL 玻璃燒杯內,加入150 mL 水,磁力攪拌5 min,靜置15 min,棄去部分上清液,剩余溶液體積約30 mL。加入與剩余溶液等體積的含鈣和鍶的乙酸溶液,蓋上表面皿,于電熱板上加熱至微沸并保持5 min,其間不時搖動。自然冷卻后,以水清洗表面皿及杯壁,而后添加與燒杯中剩余溶液等體積的乙醇,混勻,靜置30 min。以慢速定量濾紙過濾,用水洗滌燒杯及沉淀各5~6次,隨后將濾紙及沉淀轉移至鉑-金坩堝內,于100 ℃烘干,室溫入爐,爐門留縫升溫至800 ℃灰化,灼燒30 min。在所得灰分中補加二氧化硅至灰分質量達到0.800 0 g,加入8.000 0 g 混合熔劑及0.1 g 脫模劑碘化銨,于1 100 ℃熔融,靜熔300 s,擺熔300 s,靜置20 s,所得樣片按照儀器工作條件測定。
2 結果與討論
2.1 前處理方法的選擇
稀土礦石中可能影響其中目標化合物測定的干擾物質通常為硫酸鈣、碳酸鋇、碳酸鈣和碳酸鍶等,而查閱文獻[7]可知,稀土礦石不存在與毒重石(BaCO3)、菱鍶礦(Sr CO3)、石膏(CaSO4·2 H2O)等3組分礦石伴生的情況,這是由于稀土礦石中硫酸鈣與碳酸鍶、碳酸鋇或其中任一組分共存時,會在環境中水的作用下,發生置換反應生成溶度積更低的硫酸鋇和硫酸鍶,進而轉化成重晶石礦或天青石礦。因此,稀土礦石中的干擾成分可能是硫酸鈣、碳酸鈣或其中任一組分,也可能是碳酸鈣、碳酸鍶、碳酸鋇或其中任一或者任二組分。
對文獻或標準中干擾物質排除的相關方法進行總結,所得結果見表3。

表3 干擾物質排除的方法Tab.3 Method of eliminating interfering substances
由表3可知:文獻或標準方法采用了含鍶或鈣的乙酸溶液或鹽酸溶液排除干擾,其中以乙酸溶液溶解硫酸鈣、碳酸鈣、碳酸鋇和碳酸鍶;通過在乙酸溶液中添加1 g·L-1的碳酸鍶或2 g·L-1的碳酸鈣,利用Ca2+、Sr2+的同離子效應抑制目標物氟化鈣、硫酸鍶的電離;利用鹽酸溶液可有效排除硫酸鋇測定值的干擾,但其會影響氟化鈣與硫酸鍶的分離[2,4],同時,為了加速反應的進行,需于90 ℃加熱或微沸30 min。為實現3種目標化合物的測定,需按照表3中的方法對3種目標化合物進行逐一分析,但步驟繁瑣、耗費時間較長,因此有必要設計一種簡單、通用、高效的前處理方法,以排除硫酸鈣、碳酸鈣、碳酸鋇和碳酸鍶等4種物質的干擾。
綜合表3中各方法,首先選擇了含10%鈣和鍶的乙酸溶液30 mL進行兩種極端狀況(一組為硫酸鈣和碳酸鈣,另一組為碳酸鈣、碳酸鍶和碳酸鋇)下的干擾物質排除試驗,結果顯示:以質量均為0.1 g的碳酸鈣、碳酸鍶、碳酸鋇(均遠高于樣品中含量)為待測對象,微沸5 min 時,3 種干擾物均可充分溶解;以0.13 g硫酸鈣和0.1 g碳酸鈣(均遠大于樣品中對應雜質的含量)為待測對象,微沸5 min,2種干擾物質不能完全溶解,推測原因可能為乙酸溶液用量過少或乙酸溶液中添加的Ca2+抑制了硫酸鈣的溶解。
基于此,優化試驗流程如下:先以水溶解分離樣品中的硫酸鈣,考慮到樣品中硫酸鈣質量分數通常不超過10%,結合硫酸鈣在18 ℃下水中的溶解度(0.255 g),選擇加入150 mL水,通過磁力攪拌溶解樣品中硫酸鈣。由于磁力攪拌后沉淀速率較慢,且上清液與沉淀難以完全分離,為保證目標物的回收率,在棄去上清液時試驗選擇保留30 mL 溶液,但也因此稀釋了后續使用的含鍶和鈣的乙酸溶液,故將含鍶和鈣的乙酸溶液中的乙酸的體積分數提高至20%,并等比例提高其中Ca2+、Sr2+的質量濃度。結果顯示,經水處理后再以此溶液處理以上2組干擾試驗對象,干擾物均得到充分溶解,說明1.3節的前處理條件可以作為分析稀土礦石中螢石、重晶石和天青石的通用前處理方法。
2.2 熔融制樣條件的選擇
以1 100 ℃熔融樣品時,灰分可以溶解,得到的樣片清明、透亮。由于實際樣品中二氧化硅等組分呈酸性,因此試驗選擇偏堿性的熔劑組合(由質量比為12∶22的四硼酸鋰和偏硼酸鋰混合而成)進行中和,以改善熔體的流動性,保證樣品充分溶解。
在灰化結束后,為避免玻璃棒劃傷鉑-金坩堝內壁,不能采用玻璃棒混勻樣品與熔劑,試驗選擇10∶1這一相對較高的稀釋比充分分解樣品(若直接熔融樣品,可選擇在瓷坩堝中先行混勻,稀釋比可低至3∶1)。
2.3 校準曲線和檢出限
按照試驗方法分析校準用標準樣品系列,以氧化物質量分數為橫坐標,其對應的熒光強度為縱坐標繪制校準曲線,所得線性范圍、線性回歸方程和相關系數見表4。

表4 校準回歸參數Tab.4 Calibration regression parameters
本試驗涉及的樣品組成復雜、且各組分含量差異較大,待測樣品間基體效應差異明顯,所得的各氧化物的檢出限波動較大,故不對方法的檢出限進行考察。
2.4 基體效應
待測樣品組成差異顯著,元素間吸收-增強效應對樣品中目標化合物測定的影響較大,因此需校正基體效應,試驗選擇以基本參數法校正基體效應,相關計算公式見文獻[8]。
2.5 準確度與精密度試驗
將硫酸鈣、碳酸鈣、碳酸鍶、碳酸鋇與GBW 07812、氟化鈣、硫酸鍶、氧化鑭、氧化鈰、GBW 07311、GBW 07363等混合配制不同濃度水平的樣品,并按照試驗方法進行測定,結果見表5。
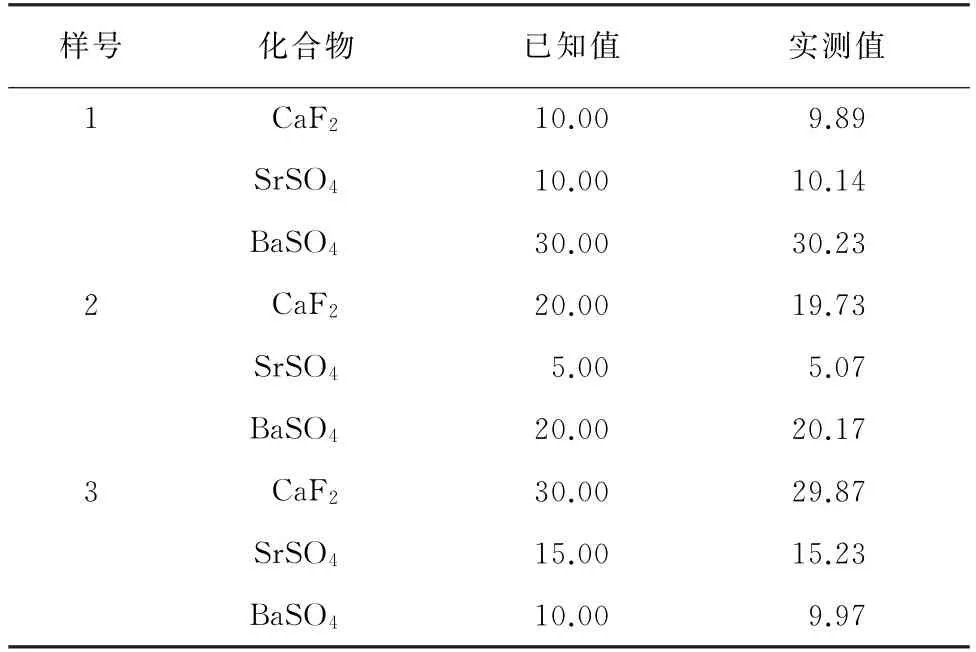
表5 準確度試驗結果Tab.5 Results of test for accurary %
由表5可知:3種化合物的測定值和已知值基本一致,說明方法的準確度較好。
按照試驗方法重復分析實際樣品6次,結果顯示:氟化鈣、硫酸鍶和硫酸鋇的檢出量分別為5.67%,21.43%,11.47%,RSD 分別為0.60%,1.7%,0.85%,說明方法的精密度較好。
本工作建立了用含鈣和鍶的乙酸溶液浸取,熔融法制樣,XRFS 同時測定稀土尾礦中螢石、重晶石、天青石等含量的方法,本方法前處理步驟簡單,試劑消耗少,所用時間短,具有一定的實用性,可推廣用于螢石-重晶石共生礦的分析領域,但是方法不能排除硅酸鹽的干擾,且僅適用于鈣、鍶、鋇硅酸鹽礦物含量較低的樣品,需要通過進一步的工作來完善。
猜你喜歡
無機鹽工業(2021年1期)2021-01-08 08:50:22
石材(2020年12期)2020-12-31 21:25:39
賀州學院學報(2020年3期)2020-02-16 08:32:02
兒童故事畫報(2019年5期)2019-05-26 14:26:14
中成藥(2018年5期)2018-06-06 03:12:18
意林原創版(2016年10期)2016-11-25 10:28:30
Coco薇(2016年2期)2016-03-22 02:42:52
廣西民族大學學報(自然科學版)(2015年3期)2015-12-07 00:56:07
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
