芳香性概念的最新進展
2021-09-26 10:50:46黃寶磊孫昊李欣燁范雨亭陶濤
大學化學 2021年8期
關鍵詞:規(guī)則
黃寶磊,孫昊,李欣燁,范雨亭,陶濤
南京信息工程大學化學與材料學院化學系,南京 210044
芳香性作為化學中最重要的概念之一,一直吸引著實驗和理論科學家的關注。芳香性并不是一個嚴格意義上的科學概念,從未被完整定義,自從這一概念被提出以來,經歷了150多年的發(fā)展,但這個古老的課題仍然有著鮮活的生命力,是有機化學中的前沿課題。作為一種概念,實驗測定和量子化學的理論計算在對芳香性的評估中均展現(xiàn)出重要作用,特別是近幾年,這一概念的內涵與外延不斷擴展。尤其在新穎芳香性的發(fā)現(xiàn)和詮釋中扮演著不可或缺的角色。
1 芳香性的研究歷史
1825年,法拉第發(fā)現(xiàn)了苯,后來人們將一類具有芳香氣味的化合物歸為“芳香化合物”,但是由于這種歸類具有主觀性、模糊性,所以并未深入研究。1855年,霍夫曼[1](August Wilhelm Hofmann)最早使用aromatic一詞表示含苯環(huán)的化合物,成為芳香性(aromaticity)的濫觴。1865年,德國有機化學家凱庫勒(Friedrich August Kekulé, 1829–1896)提出苯的結構。1890年,凱庫勒在德國化學會慶祝苯結構論文發(fā)表25周年的演講中提到,他在夢中看到蛇咬著自己的尾巴,并受此啟發(fā),想到苯分子的環(huán)狀結構。伴隨著量子化學的發(fā)展,理論化學迎來了突破。1931年,德國科學家休克爾[2](Erick Hückel,1896–1980)提出具有(4n+ 2)個π電子的平面閉合共軛單環(huán)化合物可能具有芳香性,之后合成的一系列的芳香化合物也證明了休克爾規(guī)則的正確性。凱庫勒1896年逝世,休克爾1896年出生,科學界將其稱為“苯的連續(xù)之謎”(benzene enigma continues),以此表達對兩位科學巨擘的敬仰。
1965年,Breslow[3]將(4n)個π電子的反芳香性補充到休克爾規(guī)則中。1972年,貝爾德(Colin Baird)提出當分子處于第一激發(fā)三重態(tài)時,其(反)芳香性與休克爾規(guī)則相反[4]。這兩個經典規(guī)則在芳香性的理論體系中具有劃時代的意義,指導了之后的眾多相關研究。近些年來,除了傳統(tǒng)的有機分子,金屬苯及其衍生物、其他金屬有機化合物的芳香性均有諸多報道,全局芳香性、三維芳香性、超共軛芳香性等新概念不斷涌現(xiàn)(表1)。

表1 近年來有關芳香性化合物的研究進展
2 芳香性的判據(jù)發(fā)展
化合物是否具有芳香氣味是判斷芳香化合物的起源,但是這一判據(jù)過于主觀粗糙,且不能反映這類化合物的物理、化學性質,所以逐漸被摒棄。雖然科學家對芳香性進行了深入研究并提出多種方法[12],但是最經典、被廣泛認可的是休克爾規(guī)則,并沿用至今。1931年,休克爾等人提出:在單雙鍵交替的平面環(huán)狀結構中,具有(4n+ 2)個π電子的為芳香性;后又補充為具有(4n)個π電子的為反芳香性。1964年,Heilbronner預測具有莫比烏斯環(huán)狀共軛拓撲結構的π體系中,具有(4n)個π電子的為芳香性,而不是(4n+ 2)個π電子[13],這與傳統(tǒng)的休克爾規(guī)則剛好相反,稱為莫比烏斯芳香性規(guī)則(M?bius aromaticity rule)。1972年,貝爾德提出不僅在莫比烏斯特殊的環(huán)狀條件下,當分子處于三重態(tài)時亦存在與休克爾規(guī)則相反的情況。
如何判斷化合物具有芳香性是芳香化合物被發(fā)現(xiàn)以來經久不衰的課題。芳香性是用來評價環(huán)狀共軛有機化合物具有額外穩(wěn)定性的一種概念,芳香性的形成是π電子良好重疊與π電子數(shù)合適兩方面共同決定的,前者需要以σ鍵骨架作為結構基礎,后者需要判斷具體適用于休克爾規(guī)則還是貝爾德規(guī)則。下面從實驗和理論兩個方面進行舉例闡述。
2.1 芳香性的實驗判據(jù)
核磁共振譜(NMR)仍是判斷芳香性的重要證據(jù)。苯的1H NMR譜圖中,由于所有氫原子為磁等價和化學等價,化學位移值約為7.33,而同等條件下共軛烯烴的氫原子化學位移值為4.5–6.5,主要由于存在去屏蔽效應。[18]輪烯中,12個氫原子(12H)朝外,6個氫原子(6H)朝里。以氘代四氫呋喃為溶劑,?60 °C的1H NMR譜圖中,12H化學位移值為9.25,6H化學位移值為?2.9。而在溫度為120 °C的譜圖中,所有的氫化學位移為5.45。這一實驗結果表明:(1) 高溫下,[18]輪烯朝外的氫與朝里的氫可以快速交換;(2) 芳香性作為一種理化性質,依賴溫度的變化而變化。
除了利用休克爾規(guī)則或貝爾德規(guī)則以外,科學家曾試圖通過芳環(huán)結構易取代難加成的化學反應特性、鍵長的平均化程度,以及幾何構型等角度來判斷芳香化合物,但是隨著對芳香性研究得越深入,越顯得這些判據(jù)較為粗淺。例如,富勒烯C60為足球狀化合物,其易加成的性質突破了反應特性的限制。
此外,單晶結構測定獲得分子的鍵長、鍵角等數(shù)據(jù),也可以從實驗上輔助判斷分子的芳香性。鍵長平均化程度能夠側面反映共軛的情況,置信度較高,但是其平均化程度只可與同分異構體或同系物比較,才具有實際意義。例如,環(huán)硼氮烷(B3N3H6)中,所有B-N鍵鍵長均為0.143 nm[14],而萘分子并不是等鍵長體系,但是一般認為萘比環(huán)硼氮烷更具有芳香性。目前的實驗判據(jù),均為多種結果并存分析,只依賴一種實驗結果就斷定某種新體系是否具有芳香性,顯得比較武斷。
2.2 芳香性的理論判據(jù)
實驗判據(jù)只能通過譜圖間接表現(xiàn),無法實現(xiàn)芳香性的可視化。但是隨著理論計算和圖形可視化技術的發(fā)展,這一狀況得以改善。在芳香性體系中,離域的電子在外加磁場的作用下會產生明顯的感應電流,因此研究分子的電磁性質是研究芳香性的有效手段。通過繪制矢量圖,可以直觀地觀察到分子內感應電流,從而可以判斷分子有無芳香性。其中,各向異性感應電流密度和核獨立化學位移的優(yōu)勢逐步顯現(xiàn),成為主流的理論判據(jù)。
各向異性感應電流密度(anisotropy of the induced current density,AICD[15])本質上反映的是相應位置電子對磁場感應的各向異性的強度。其中,環(huán)電流方向與左手規(guī)則相同時,電流越大則芳香性越強;而電流方向越與左手規(guī)則相反時,電流越大則反芳香性越強;如果沒有產生明顯凈電流,則是非芳香性。
核獨立化學位移(nucleus-independent chemical shift,NICS[16])是被使用得最廣泛的衡量芳香性的指標,其含義為設定的不在原子核位置上的磁屏蔽值的負值,負值越大,表明對磁場屏蔽越強,則芳香性越強。一般來說,如圖1所示,設定的位置取在共軛環(huán)的幾何中心,被稱為NICS(0)。更進一步,在實際計算中有兩種NICS值:各向同性值NICS(0)iso(磁屏蔽張量xx、yy、zz之和的平均)和磁屏蔽張量zz分量值NICS(0)zz(z軸垂直于環(huán)平面)。
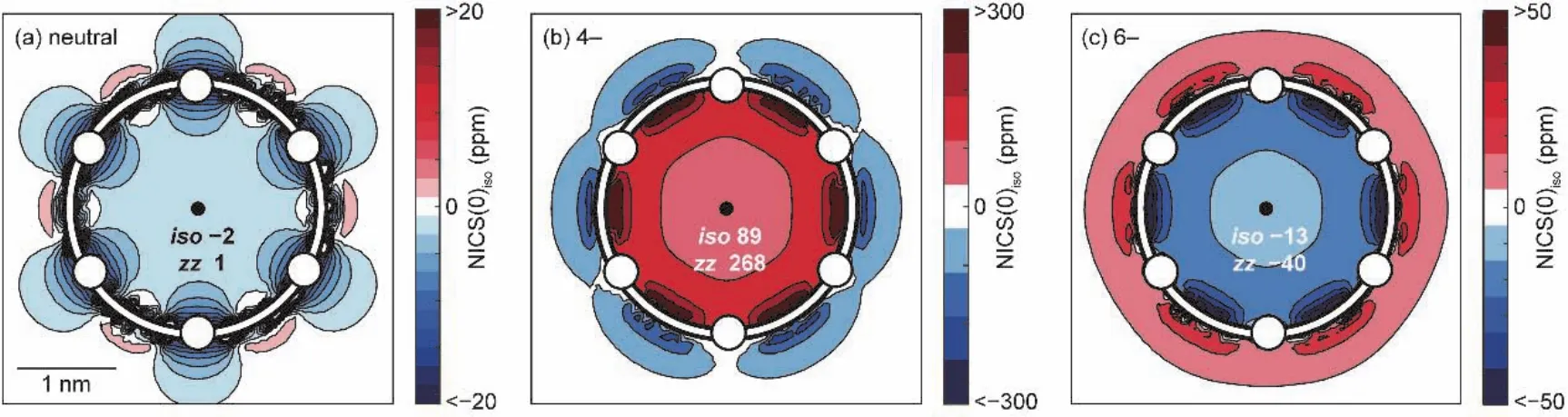
圖1 卟啉納米環(huán)NICS計算結果示意圖
3 芳香性的最新進展
隨著研究的深入,芳香性這一性質已經從起初的苯環(huán)衍生物,到現(xiàn)在體現(xiàn)在了不同類型的化合物上,包括卟啉類化合物、金屬雜環(huán)化合物、無機金屬團簇等;描述其規(guī)律的理論規(guī)則也在不斷發(fā)展,逐漸趨于體系化,立體芳香性、全局芳香性等新的芳香性概念紛紛涌現(xiàn)。
近年來,社會互聯(lián)網化進程不斷加快,電子商務創(chuàng)新不斷涌現(xiàn),電子商務企業(yè)對傳統(tǒng)商貿企業(yè)形成了激烈的跨界競爭態(tài)勢。對于重慶市商貿流通業(yè)而言,順應互聯(lián)網時代要求,實現(xiàn)向互聯(lián)網化的轉變,不僅要加快構建線上銷售服務能力,實現(xiàn)業(yè)務上網,以滿足消費者日益增長的在線消費需求,更要發(fā)揮已有的傳統(tǒng)線下優(yōu)勢,將線下資源與線上業(yè)務相互結合,形成線上線下互動創(chuàng)新發(fā)展的新興商業(yè)模式。
3.1 立體芳香性
立體芳香性,也稱三維芳香性,即三維空間上的芳香性。傳統(tǒng)休克爾規(guī)則適用于平面芳香性,但是自C60分子被發(fā)現(xiàn)以來,芳香性的界限已經被拓寬到三維空間上,隨著近年來莫比烏斯體系化合物合成技術的進步,立體空間上的芳香性逐漸頻繁地出現(xiàn)在人們的視線之中。
1858年莫比烏斯等人提出莫比烏斯環(huán)的概念,雖然自然界沒有莫比烏斯型分子,但是化學家一直試圖去合成這類分子,其難度可想而知。2009年Kim課題組[17]總結了拓展型卟啉類化合物中的莫比烏斯芳香性和反芳香性,如果實現(xiàn)莫比烏斯環(huán)狀芳香性,需要構象柔性好、翻轉能力強、氧化還原活性高和具有潛在配位點等優(yōu)勢。如圖2所示,在具有28個π電子的拓展卟啉結構中呈現(xiàn)出芳香性,這與休克爾規(guī)則恰好相反。
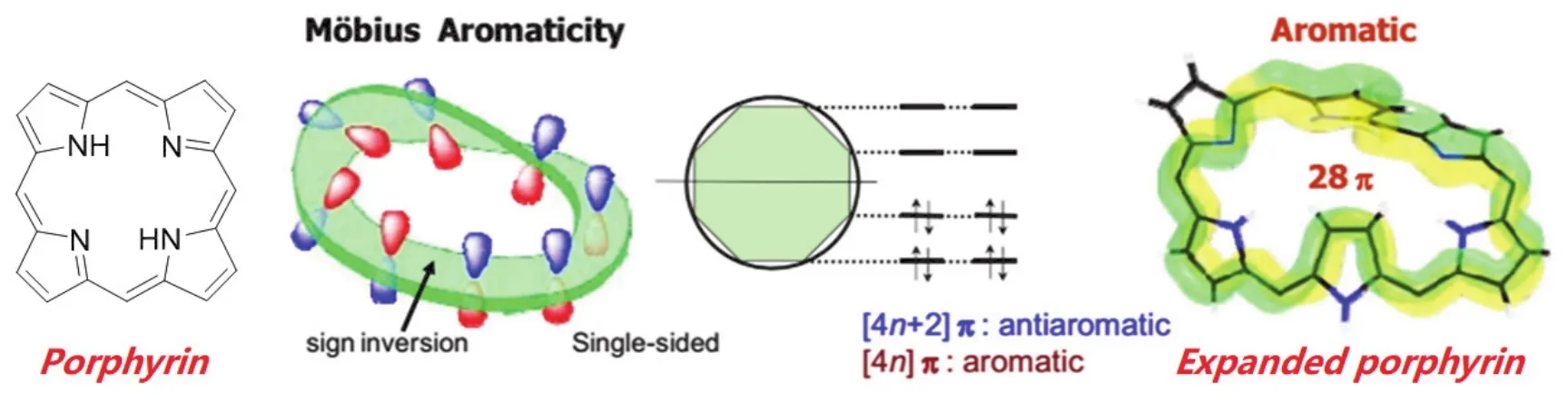
圖2 拓展型卟啉類化合物中莫比烏斯芳香性和反芳香性[17]
既然莫比烏斯環(huán)的芳香性與平面芳香性不同,那么其他的立體結構芳香性又將如何?這一點引起了科學家持續(xù)不斷的探索,而不僅拘泥于莫比烏斯型結構。2017年Kim團隊[6]報道了并三噻吩橋聯(lián)的共軛大環(huán)化合物1a和2a (圖3),兩者的區(qū)別在于1a的大環(huán)結構拓展單元為雙噻吩,而2a中為并三噻吩,在同一個框架內有多個電子回路并且π電子可被不同的電路共同占有,是一種雙環(huán)共軛體系。在中性狀態(tài)中,形成了34個π電子和26個π電子兩個相互競爭的芳環(huán)體系;在失去兩個電子的氧化態(tài)中,[4n+ 1]/[4n+ 1]雙自由基三重態(tài)顯示出全局芳香性,即33個π電子和25個π電子的芳環(huán)體系,除去共用電子,總計40個π電子恰好符合貝爾德規(guī)則。

圖3 噻吩類共軛大環(huán)化合物的立體芳香性[6,10]
2020年吳繼善課題組[10]非常巧妙地通過Friedel-Crafts烷基化和Yamamoto偶聯(lián)反應合成了一種含有12個噻吩結構的完全共軛的雙自由基分子籠c-T12,可根據(jù)對稱性、π電子數(shù)目和自旋態(tài)獲得不同類型的芳香性。在[38π]中性狀態(tài)下為單環(huán)共軛休克爾芳香性;在+2氧化態(tài)下為單環(huán)共軛貝爾德芳香性;在+4氧化態(tài)下為多環(huán)共軛立體全局反芳香性;在+6氧化態(tài)下為多環(huán)共軛立體全局芳香性。
如圖3所示,兩種多環(huán)共軛分子通過改變自身價態(tài),不僅可改變芳香性的程度,而且可改變芳香性的種類,特別是分子c-T12,通過改變分子價態(tài)來改變甲基上氫質子在低場與高場中的指向,實現(xiàn)分子籠立體反芳香性與芳香性之間的轉換,這些工作是芳香性概念發(fā)展中的里程碑。
3.2 全局芳香性
全局芳香性與立體芳香性既有聯(lián)系又有區(qū)別,立體芳香性是針對平面芳香性而言,全局芳香性的概念是建立在環(huán)電流磁學判據(jù)的基礎上提出的,針對局部芳香性,因而其研究對象多為具有多個基本單元的大環(huán)化合物,其中最典型是苯環(huán)類大環(huán)多自由基化合物和卟啉納米環(huán)。
3.2.1 苯環(huán)類大環(huán)多自由基化合物中的全局芳香性
迄今為止,卟啉類化合物中已經出現(xiàn)了很多大環(huán)(反)芳香族化合物,但是由于苯環(huán)的π電子存在較大的共振能量,使得苯環(huán)類大環(huán)化合物無法表現(xiàn)出全局芳香性,因此具有全局芳香性的苯環(huán)類多環(huán)芳烴很少被合成。值得注意的是,當分子處于開殼雙自由基或多自由基的形式時,它們可以適應自身的幾何形狀和電子自旋狀態(tài)以達到最低能態(tài),趨于芳香化。

圖4 凱庫勒烯共振雜化體3a和3b [18]

圖5 系列大環(huán)多自由基化合物[19]
這些大環(huán)多自由基分子有諸多共同點,它們普遍存在兩種或兩種以上共振結構,在共振中存在著芳香六元環(huán)的形成與破裂,并且這些分子處于一種局部芳香性與全局(反)芳香性的平衡狀態(tài),但全局(反)芳香性往往占主導作用,可以用休克爾規(guī)則去解釋他們的芳香性,卻又不完全受限于休克爾規(guī)則。并且?guī)缀跛械倪@種類型的大環(huán)結構都能夠通過在環(huán)戊環(huán)的碳位點上添加額外的質子巧妙地實現(xiàn)全局芳香性與局部芳香性之間的轉換,由此引發(fā)我們對局部芳香性與全局芳香性之間的轉化問題的探討。
最值得一提的是,吳繼善課題組[7]合成了一類五元環(huán)和六元環(huán)交替稠合的大環(huán)多自由基化合物——具有碗狀骨架8MC和近乎平面骨架10MC,在這種結構中內環(huán)與外環(huán)的行為相互獨立(圖6)。在碗狀分子8MC中,內環(huán)有24個π電子,外環(huán)有32個π電子,處于基態(tài)的外環(huán)與內環(huán)皆有[4n]個π電子,然而根據(jù)1H NMR顯示外環(huán)氫質子位于低場(δ= 11.37),內環(huán)氫質子位于高場(δ= ?12.08):表明該化合物具有很強的芳香性,這與休克爾規(guī)則恰恰相反。對于10MC來說,內環(huán)和外環(huán)分別具有30和40個π電子,內環(huán)為單重態(tài)滿足休克爾規(guī)則,而外環(huán)為三重態(tài)遵循貝爾德規(guī)則,對于整個大環(huán)來說具有全局芳香性的特征。對于8MC的不尋常現(xiàn)象,作者認為在8MC中內環(huán)與外環(huán)都具有三重態(tài)雙自由基的特征,而兩個三重態(tài)環(huán)耦合為一個單重態(tài)大環(huán)即“內三重態(tài)-外三重態(tài)”是這種全局芳香性形成的原因。這些研究為輪烯套輪烯結構的設計提供了合理借鑒。
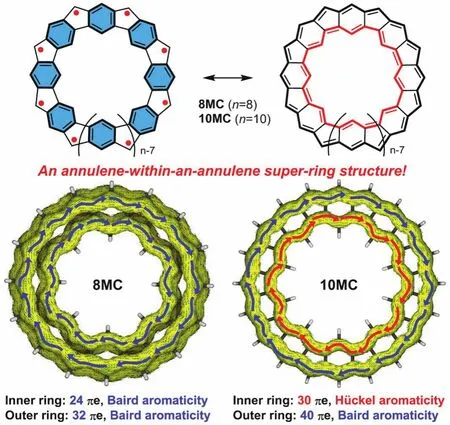
圖6 具有輪烯套輪烯結構和全局芳香性的大環(huán)多自由基[7]
3.2.2 卟啉納米環(huán)在全局芳香性中的研究
卟啉是一具有代表性的物質,據(jù)研究表明,它的π電子可以多達162個,周長可以達到16 nm[20],屬于超分子環(huán),在大環(huán)芳香性的研究中是不可或缺的,尤其是對研究大環(huán)芳香族化合物芳香性轉換具有重要意義。卟啉納米環(huán)具有優(yōu)異的性質,它可以通過改變氧化態(tài)、還原態(tài)和基本組成單元的數(shù)目實現(xiàn)全局芳香性和全局反芳香性的轉換,這對預測激發(fā)態(tài)的行為具有重要意義。
如圖7所示,Martin D. Peeks和Michael Jirasek等人報道了六卟啉納米環(huán),分別為c-P6[20]和c-P6·T6[9]。通過改變部分卟啉基本單元氧化態(tài)來改變整體大環(huán)的氧化態(tài)從而實現(xiàn)改變芳香性的目的。由于中性狀態(tài)下每個卟啉分子都能維持獨立的環(huán)電流,因此卟啉類大環(huán)在中性時僅表現(xiàn)出局部芳香性,并不表現(xiàn)出全局(反)芳香性。對于c-P6·T6分子,當其失去4個電子時(即分子呈現(xiàn)+4價,80個π電子)具有全局反芳香性,當其失去6個電子時(即分子呈現(xiàn)+6價,78個π電子)具有全局芳香性。隨后通過增加卟啉環(huán)的電子個數(shù)的方式來改變它的芳香性,發(fā)現(xiàn)卟啉環(huán)的兩種還原態(tài)?4價(88個π電子)和?6價(90個π電子)與氧化態(tài)相似,分別表現(xiàn)出全局反芳香性和全局芳香性,符合休克爾規(guī)則。
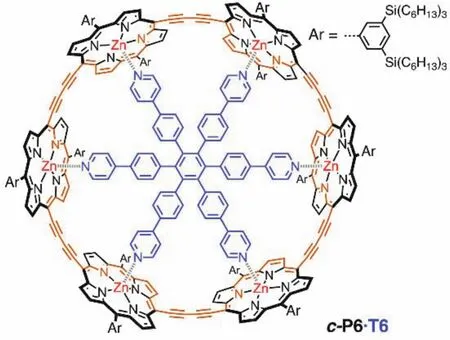
圖7 卟啉納米環(huán)中的全局芳香性和反芳香性[9]
Harry L. Anderson教授團隊成功合成了具有162個π電子的卟啉納米環(huán),這是史上最大的芳香環(huán)[11](圖8)。通過與不同的模板分子結合可以改變環(huán)的幾何結構。NMR結果顯示:當環(huán)幾何結構為扭曲8字形時無全局環(huán)電流,這表明通過改變分子的幾何結構,芳香性可以被巧妙地打開或關閉。這一結論為可芳香性分子開關的研究提出了新的解決方案。
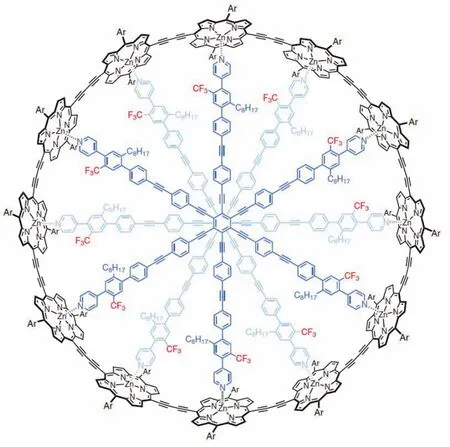
圖8 目前最大芳香納米環(huán)c-P12[b12]·(T6ef)2 [11]
除氧化還原和幾何結構,電子自旋態(tài)的改變對芳香性亦有影響。激發(fā)態(tài)卟啉納米環(huán)(反)芳香性較難被實驗證明,因為NMR譜圖并不適用于激發(fā)態(tài),而大環(huán)的NICS值也因尺寸和不對稱性受限。將紫外-可見吸收光譜和紅外光譜用于激發(fā)態(tài)下大環(huán)化合物芳香性的評估:(1) 在用紫外吸收光譜檢測時,反芳香性卟啉的三重激發(fā)態(tài)的光譜峰型顯得寬而離散,不如芳香性卟啉的尖銳而有特征,類似芳香化合物基態(tài)的吸收光譜形狀,但最近實驗表明反芳香性卟啉也能展現(xiàn)尖銳的峰型,因研究過少無法證明其正確性;(2) 在用紅外光譜檢測卟啉單重態(tài)激發(fā)態(tài)的芳香性時,更少的紅外活性振動對芳香性分子的對稱做出了很好的解釋;(3) 大環(huán)環(huán)電流的大小可以在某種程度上被拓撲結構所控制的,這在小環(huán)里是根本無法體現(xiàn)的。
總之,含有5–9個卟啉單元的納米環(huán)含有70–126個π電子,已經超出經典的貝爾德規(guī)則范疇,但其第一激發(fā)三重態(tài)的芳香性經過密度泛函理論DFT計算證明,仍然符合貝爾德規(guī)則。例如162個π電子的大環(huán),這些超分子環(huán)的結構仍然符合休克爾規(guī)則。與此同時,也存在諸多新問題:(1) 當納米環(huán)在還原態(tài)和氧化態(tài)同時具有全局芳香性時,處于還原態(tài)的納米環(huán)的全局芳香性是否一定比處于氧化態(tài)的納米環(huán)的全局芳香性表現(xiàn)得更為明顯?(2) 通過失去電子來實現(xiàn)全局芳香性的化合物是否也可以通過得到電子來達到這一效果?(3) 納米環(huán)局部芳香性和全局芳香性切換的分子開關機制是什么?這些科學問題,都有待進一步理解芳香性的概念及芳香性強弱的影響因素。
3.3 其他芳香性
3.3.1 超共軛芳香性
1999年,理論化學家Schleyer和Nyulászi[21]通過計算化學發(fā)現(xiàn),在環(huán)戊二烯中sp3雜化碳原子上兩個給電子取代基可以通過超共軛作用參與環(huán)內的π共軛,形成超共軛芳香性。2016年,清華大學趙亮團隊與廈門大學朱軍團隊合作首次報道了以過渡金屬作為取代基的吲哚四核金簇中存在超共軛芳香性[5],同時他們發(fā)現(xiàn)過渡金屬取代基比傳統(tǒng)主族元素作為給電子取代基可以產生更為顯著的超共軛芳香性效應。
3.3.2 自適應芳香性
根據(jù)貝爾德法則,一般情況下芳香性分子在第一激發(fā)三重態(tài)下會出現(xiàn)與基態(tài)相反的芳香性,2018年朱軍等人合成了一種有機金屬化合物[22](圖9),此分子在S0和T1狀態(tài)下同時具有芳香性。根據(jù)NICS計算、前線軌道分析和電子自旋密度的芳香性分析都得出了相同的結論:該分子在中性時與休克爾和貝爾德法則相吻合,即S0態(tài)下具有芳香性,T1態(tài)下具有反芳香性,而其陽離子狀態(tài)時卻得出與以往規(guī)律不同的現(xiàn)象:在S0和T1態(tài)下都具有芳香性,將之稱為自適應芳香性。研究認為平面內的激發(fā)比平面外的π–π*作用對形成自適應芳香性更有利,此研究將過渡金屬用于芳香性分子的研究中,為之后的自適應芳香性的研究和其他芳香性分子的合成提供了合理的借鑒。
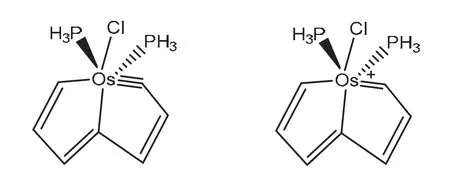
圖9 鋨雜戊搭烯S0和T1態(tài)的自適應芳香性[21]
4 芳香性的未來應用
由于芳香體系有獨特的穩(wěn)定性,眾多涉及芳構化的反應擁有一些特殊的芳香驅動力[14],在光電材料領域有長足的應用。例如激發(fā)態(tài)芳香性的理論信息仍處于匱乏狀態(tài),但可用于解釋光化學反應的合理性,解釋液晶中的光誘導結構變化[23]。在光學材料方面,三維球形芳香性的發(fā)展也帶來了不一樣的思路,提出了混合共軛π共軛橋的概念。大環(huán)芳香性化合物有著很多優(yōu)異的性能,例如大環(huán)多自由基化合物的小帶隙可以很好地穩(wěn)定多個電荷。由于離域電子波函數(shù)之間相互干涉,因此更大的芳香環(huán)可能會產生不尋常的量子效應。巨大芳香環(huán)為制造超導電流回路提供新方法,有望成為微納環(huán)超導材料,在量子計算機的組成上有潛在的應用價值。
5 結語
本文綜述了芳香性概念的最新進展,從研究背景到理論與實驗的判據(jù),特別是近五年來,大環(huán)化合物全局芳香性的研究,討論了不同氧化態(tài)、不同激發(fā)態(tài)、不同幾何結構對芳香性強弱、全局芳香性與局部芳香性之間的轉換以及全局(反)芳香性之間的切換的影響,見證了休克爾和貝爾德規(guī)則對各種超出預期范圍的大環(huán)化合物強大的理論包容性,同時看到了芳香性這一概念的歷久彌新。
猜你喜歡
作文周刊·小學一年級版(2022年28期)2022-05-30 10:48:04
小獼猴智力畫刊(2022年3期)2022-03-29 01:09:42
數(shù)學小靈通(1-2年級)(2021年4期)2021-06-09 06:26:14
法律方法(2019年3期)2019-09-11 06:26:16
中國外匯(2019年7期)2019-07-13 05:44:52
幸福(2018年33期)2018-12-05 05:22:42
環(huán)球飛行(2018年7期)2018-06-27 07:26:14
Coco薇(2017年11期)2018-01-03 20:59:57
暨南學報(哲學社會科學版)(2016年9期)2017-01-15 13:52:02
運動(2016年6期)2016-12-01 06:33:42
