教材中價層電子對互斥理論的問題探討
2021-09-26 10:51:12李保山
大學化學 2021年8期
關鍵詞:方法
李保山
北京化工大學化學學院,北京 100029
N. V. Sidgwick等把原子結構中的保利不相容原理,即“自旋相同的電子必須遠離”這一原則推廣至分子結構中,提出在一些簡單的共價分子(ABn)中,中心原子A的價層軌道電子對是環繞中心原子A排列成對稱的幾何結構的,這種結構使得各電子對相互遠離,電子對之間的凈斥力達到最小,分子體系的能量達到最低。據此,可以推測這些簡單分子中中心原子價電子對的幾何分布情況,進一步推測分子的幾何構型。這種定性解釋分子幾何構型的近似方法稱之為“價層電子對互斥理論(Valence Shell Electron Pair Repulsion Theory)”,簡稱VSEPR法。
VSEPR法比較簡單,易于理解和掌握[1–16]。對于絕大多數ABn型的簡單共價分子、離子的幾何構型均可以準確地推測,可以認為這一方法在認識這些簡單共價分子、離子的幾何構型方面上升到了比雜化軌道理論更科學的層面,因為雜化軌道理論只能解釋已知幾何構型的簡單共價分子或離子的幾何結構,而不能推測其幾何結構。因此,VSEPR法得到了廣泛的運用。
但在目前的教材中對于VSEPR法的理解尚不統一,其應用方法存在著多個版本,有些教材對這一理論應用的描述難以理解,有的添加了更多的假定成分,易于導致初學者產生疑惑。
本文對目前國內外常見的幾種教材對這一理論的應用過程進行對比分析,旨在形成對這一化學基本理論的共識。
1 VSEPR法理論要點
在利用VSEPR法推測簡單分子(或離子)中中心原子的價電子幾何分布情況時,可以將這些分子(或離子)記為ABnEm。其中A為中心原子,B為配位原子,E為孤對電子,n和m分別是成鍵電子對數和孤電子對數。該理論的基本要點[1–10]:
1) 中心原子形成的共價鍵數與原子的價層軌道數及價電子數有關,多數原子在形成分子時已經成對的電子可能會被激發到能量較高的p軌道或d軌道上,從而形成更多的價層軌道,所以中心原子成鍵時可以不受八隅律限制,其價層電子對數可能大于4 (即n+m大于4),甚至可達12 (表1)。
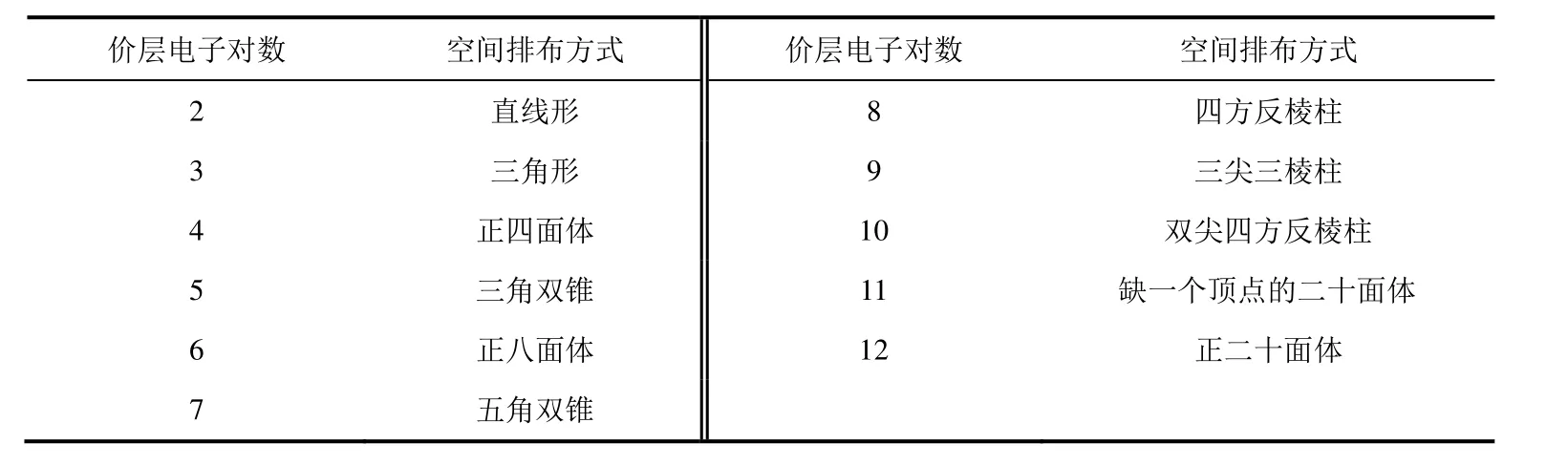
表1 價層電子對的高對稱性空間排布方式
2) 中心原子A的價電子層(即最外層)電子對彼此排斥,盡可能地遠離,最終形成高度對稱的空間排布結構。
價層電子對具有兩種類型:成鍵的σ鍵電子對(骨架電子對,用BP表示)和沒成鍵的孤電子對(用LP表示)。分子中除了電子對間的排斥力之外,所有電子對還都受A、B原子核的吸引力,當這兩種力達到平衡時,就決定了價層電子對的位置,價層電子對在中心原子周圍的分布情況與價層電子對數目有關。常見的電子對高對稱性空間排布方式見表1。
3) 價層電子對之間排斥力的大小取決于電子對在中心原子周圍分布的夾角和電子對的類型。排斥力大小順序為:
① LP-LP > LP-BP > BP-BP。
② 三鍵>雙鍵>單鍵。
可見價層電子對間夾角的大小順序為:LP-LP > LP-BP > BP-BP。
4) 分子或離子的幾何結構主要取決于中心原子A與配位原子B之間形成的σ鍵,π鍵對分子骨架結構的影響較小,其存在的位置對相鄰的價層電子對具有較大的斥力,進而對鍵角產生一定的影響。
5) 配位原子的電負性也會影響鍵角的大小。電負性越大,σ鍵電子對越遠離中心原子,受其他電子對的斥力就越小,進而對鍵角產生一定的影響。
可見,分子或離子的幾何結構主要取決于中心原子價層電子對數和價層電子對的類型,π鍵及配位原子的電負性僅對鍵角產生較小的影響。因此,可以用這一理論定性地推測簡單共價分子或離子的幾何結構。
但目前不同教材中對這一理論的應用方法介紹不統一,主要是確定中心原子的價層電子對數方法不統一,導致教學過程中出現了一些問題。下面就目前常用的幾種教材中確定中心原子的價層電子對數方法作一介紹。
2 確定中心原子價層電子對數的主要方法
2.1 根據Lewis電子式確定
這種方法是先要寫出分子或離子的Lewis電子結構式,判斷骨架電子對數和孤電子對數,進一步判斷分子或離子的幾何結構[1–4]。該方法基于八隅律,例如電子式為,中心原子S的價層有3對成鍵電子和一對孤電子,中心原子的價層電子對數為4。再如,要滿足C、O原子的八隅結構,C與O原子之間要形成雙鍵,所以C原子的骨架電子對數為3。同樣,SO3的Lewis電子式可寫為,要滿足S、O原子的八隅結構,S與其中一個O原子之間要形成雙鍵,所以S原子的骨架電子對數為3。
這種方法雖然比較直觀,但方法的適應性不強[8]。
2.2 根據中心原子的族數及配位原子提供的電子數確定
國內一些教材[5–12]給出的價層電子對數VP的計算公式為:
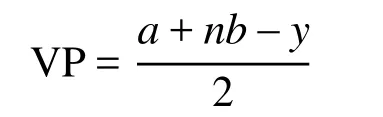
上式中a為中心原子A的價電子數,n為配位原子數,b為配位原子提供的電子總數;y為離子的電荷數(代數值),陽離子為“+”,陰離子為“?”。若出現小數(如0.5、1.5等),則進為整數(1或2)即可。該式的物理意義可以理解為:中心原子周圍的總價層電子數等于中心原子的基態價層電子數和配位原子提供的價層電子數之和。
這些教材中認為:作為配位原子的B原子通常是氫、鹵素、氧和硫。在計算配位原子提供的電子數時,氫和鹵素記為“1”,氧和硫記為“0”,即認為氧和硫作為配位原子時,不提供價電子,而氧和硫作為中心原子時,價電子數記為“6”[5–8]。
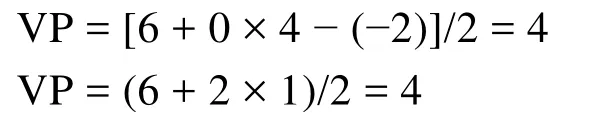
再如BrF5和XeOF4中原子Br和Xe的價層電子對數分別為:
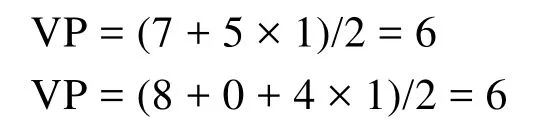
這種方法可以推測簡單分子的幾何結構,但這些教材[5–12]中認為:氧和硫作為配位原子時提供的電子數為0,而作為中心原子時提供的價電子數為6。這樣的處理方法對于初學者而言是不易理解和接受的,因為這樣的說法實際上不準確。
正確的描述應該是:“氧和硫作為端原子配位(即除中心原子外無其他原子與其鍵連)時不提供價電子”,而不是作為配位原子時不提供價電子。因為氧和硫為端原子時,是通過雙鍵與中心原子配位相連的,而π鍵對分子骨架結構的影響較小,若氧或硫原子按提供2個價電子計算,結果中將多出π鍵的1對電子,為了避免這種情況,規定:氧和硫為端原子配體時不提供價電子。若氧和硫作為端基配位原子而非端原子配位時,如―OH,就不能認為O原子不提供價電子,而是提供1個價電子。
2.3 根據基態中心原子的價電子數及配位原子的未成對電子數確定
該方法給出中心原子A的價電子對數的計算公式為[13–16]:
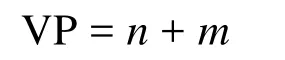
式中n和m分別為A與B之間形成的σ鍵數和孤電子對數,其中m的計算公式為:
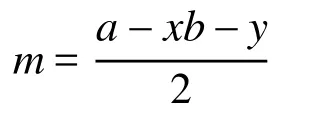
式中a為中心原子的基態價電子數,b為配位原子的未成電子數,x為配位原子數,y為離子的電荷數(代數值)。若出現小數(如0.5、1.5等),則進為整數(1或2)即可。該式具有明確的物理意義,即孤電子對數等于中心原子基態價層電子數與配位原子和中心原子之間的成鍵電子數(包括σ鍵和π鍵)之差的1/2。可以理解為配位原子B的未成對電子均要與中心原子的電子配對,即B原子也要滿足八隅律。
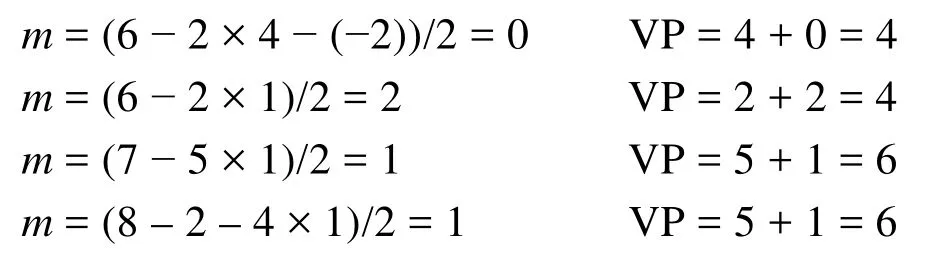
需要說明的是,配位原子的未成電子數b是指其未與中心原子成鍵的電子數。若配位原子為端原子,其未成對電子數等于其基態的未成對電子數;對于非端原子(即端基)配位,其未成對電子數應該減去與其他原子鍵連所用去的電子數,若與其他原子以單鍵相連則減去1,雙鍵減去2,三鍵減去3。例如中的端原子O,提供的未成對電子數為2,而端基H3C―和―O―CH3中的配位原子C及O的未成對電子數均為1。這是因為端基中C的4個未成對電子中已有3個與氫原子鍵連,而氧的2個未成對電子中有1個與端基―O―CH3中的C鍵連。再如,與中心C*原子相連的1位C有3個電子已與其他原子配對,2位C有2個電子已與其他原子配對,所以1位和2位上的C提供的未成對電子數分別為1和2。
這種計算方法沒有規定某原子提供的電子數是多少,比2.2節的方法易于理解和掌握。
2.4 根據相關原子的基態價層電子總數及配位原子的八隅律確定
該方法計算中心原子A的價電子對數的計算公式與2.3節方法一致[17–20]:
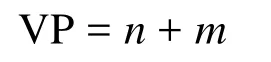
式中n和m分別為A與B之間形成的σ鍵數和孤電子對數,其中m的計算公式為:

式中a為中心原子A的基態價層電子數,n為配位原子數,b為配位原子的基態價層電子數,n1和n2分別為配位的非氫原子數和氫原子數,y為離子所帶電荷的代數值。這個公式的物理意義與2.3節的公式基本相同,可以理解為配位原子均形成了八隅體(氫原子除外),中心原子周圍的孤電子數等于總的價層電子數減去配位原子與中心原子的成鍵電子數。
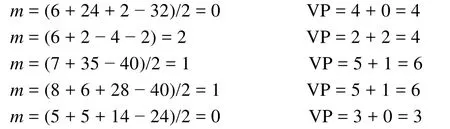
這種方法與2.3節的方法均是以基態原子的電子結構及配位原子的八隅律(氫原子除外)為基礎,沒有其他假定,且均可獲得滿意的推測結果,易于理解和掌握。相比之下,作者認為2.3節的方法更簡便、易懂、易記。
3 分子或離子幾何結構的推測方法步驟
價層電子對在中心原子周圍的幾何分布情況,就決定了分子或離子的幾何構型。
對于孤電子對數等于零的情況,分子或離子的幾何構型與價層電子對構型一致;而對于孤電子對數不等于零的情況,要通過不同類型的電子對之間的排斥力大小來排布配位原子的位置。
當分子中有π鍵存在時,π鍵應排在相當于孤對電子的位置。另外,π鍵和配位原子電負性也會使鍵角發生變化。
下面給出推測分子或離子幾何結構的方法步驟。
第一步:確定中心原子及配位原子。
一般認為分子(或離子)中氧化數絕對值最大者為中心原子,和中心原子直接相連的其他原子為配位原子(包括端原子和端基配位原子)。也可以指定分子中的某個原子為中心原子,與其直接相連其他原子或原子團為端原子或端基。但F、H幾乎均為配位原子,這是因為F的電負性最大,而氫原子只有1個單電子。
第二步:以第2.3節方法為例計算孤電子對數和VP數。
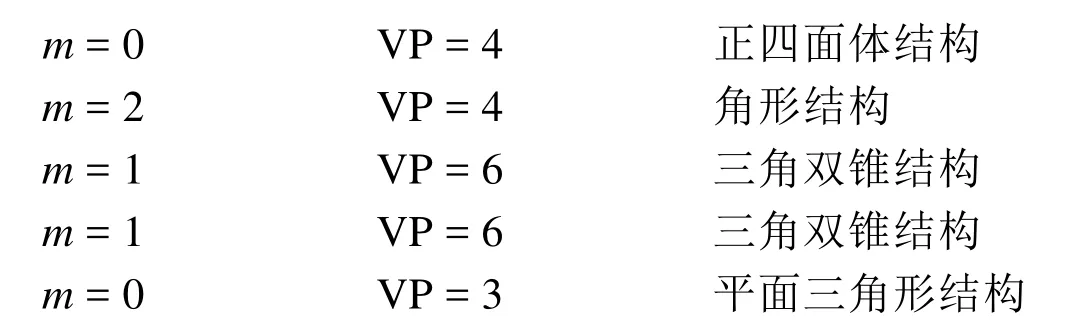
當配位原子上連接除中心原子以外的其他原子時,其未成對電子數應該減去配對其他原子的電子數,與其他原子若以單鍵相連則減去1,雙鍵減去2,三鍵減去3。
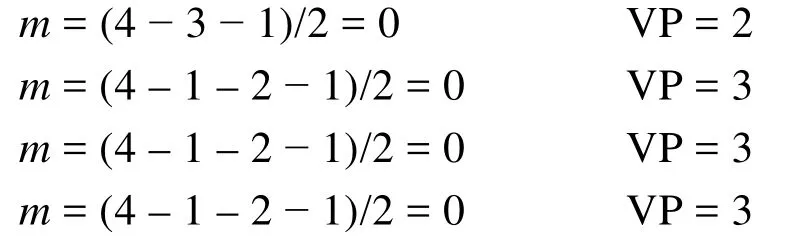
第三步:推測分子或離子的結構。
1) 孤電子對數等于零的分子或離子,其幾何構型與價層電子對構型一致。但當分子中有π鍵存在時,π鍵應排在相當于孤對電子的位置。表2給出了推測這類分子結構的一些實例。
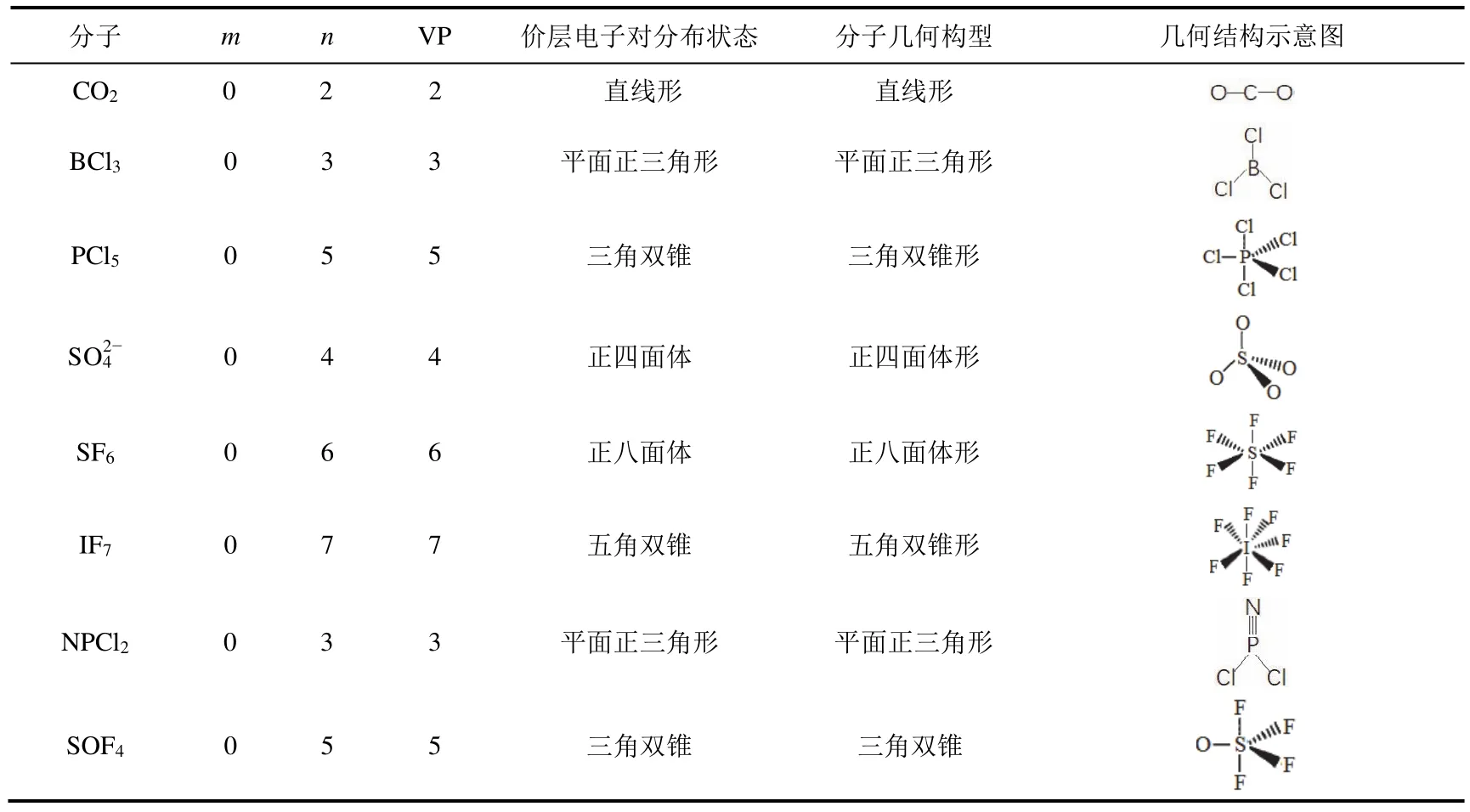
表2 孤電子對數等于零的分子結構推測實例
2) 孤電子對數不等于零的分子或離子,其幾何構型與價層電子對構型不一致,需要通過不同類型的電子對之間的排斥力大小來排布配位原子的位置。表3給出了推測這類分子結構的一些實例。
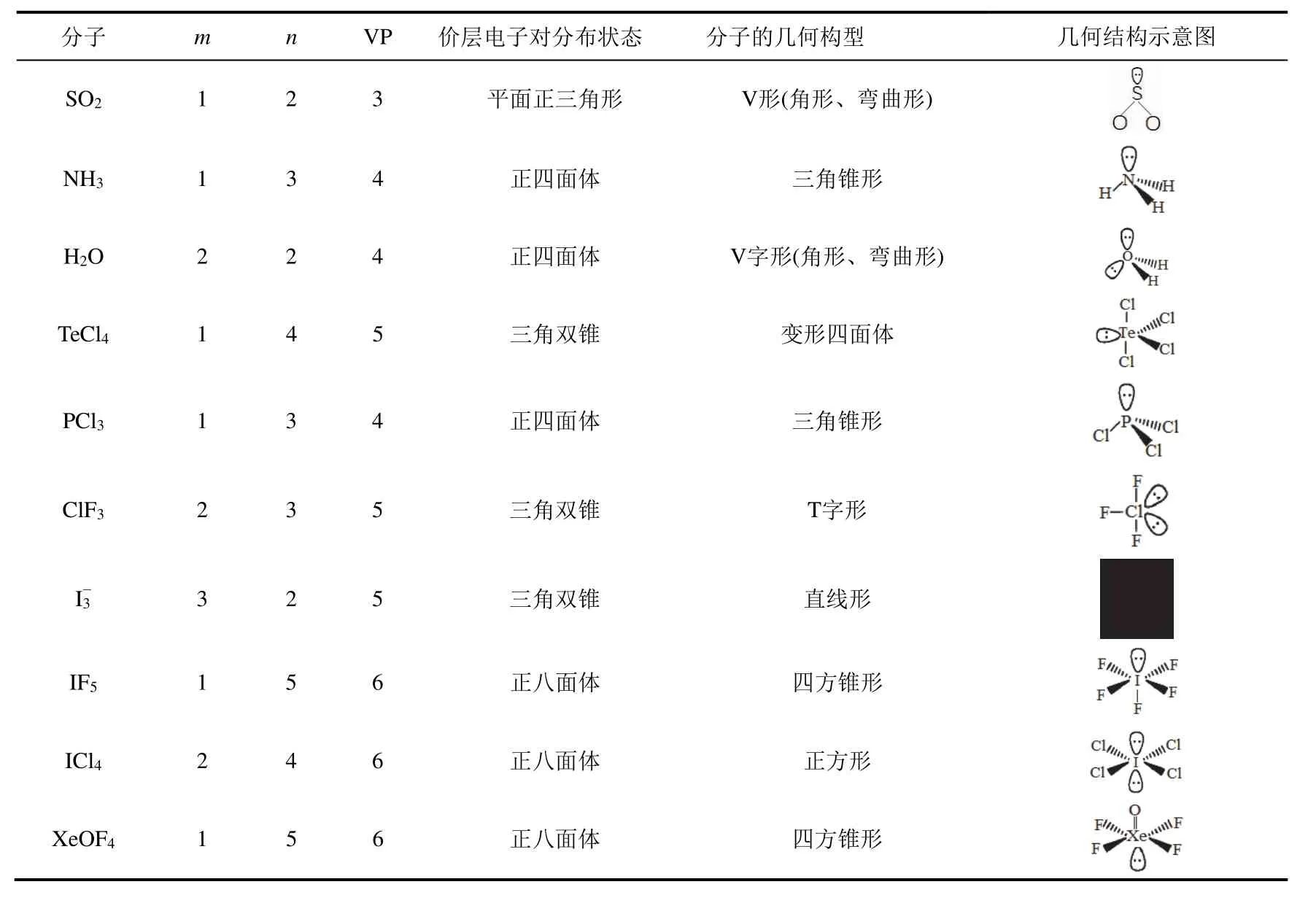
表3 孤電子對數不等于零的分子結構推測實例
3) 孤對電子、π鍵和配位原子的電負性會對鍵角產生一定的影響。孤對電子距中心原子近,對相鄰的σ鍵電子對具有較大的排斥力。同樣,π鍵電子也對相鄰的σ鍵電子對也具有較大的排斥力。這種較大的排斥力將會使臨近的σ鍵角變小;中心原子及配位原子的電負性也會使鍵角發生一定程度的變化。當配位原子的電負性較大時,由其形成的σ鍵電子對距離中心原子較遠,σ鍵電子對之間的排斥力減小,導致鍵角減小;反過來,當中心原子的電負性較大時,由其形成的σ鍵電子對距離中心原子較近,σ鍵電子對之間的排斥力較大,導致鍵角增大。
價層電子對互斥理論能夠定性的地推測簡單分子的幾何結構,方法簡單、方便,易于理解和掌握。
4 結語
通過對不同教材中關于價層電子對互斥理論的應用方法的分析及對比,雖都能定性地推測出簡單分子的幾何結構,但有的方法易于理解和掌握,而有的方法不易理解。推薦用中心原子的基態價電子數及配位原子的未成對電子數來確定中心原子價層中的非鍵電子對數(孤電子對數),該方法既具有明確的物理意義,又能夠準確解釋簡單分子或離子的幾何結構,且簡單、易懂、易記,有利于初學者的理解和掌握。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
