淺談物理化學中過渡態的搜索方法
2021-09-26 10:51:16張欣賀培楠
大學化學 2021年8期
張欣,賀培楠
北京化工大學化學學院,化工資源有效利用國家重點實驗室,北京 100029
1 引言
物理化學是化學類專業學生本科階段的一門學位課程。化學動力學是研究化學反應中速率和機理的理論[1],是物理化學學習中的重要內容。化學動力學的發展先后經歷了碰撞理論、過渡態理論和單分子反應理論等。其中,過渡態理論是目前研究最多、應用范圍最廣的化學動力學理論。對過渡態所處的“舊鍵未完全斷裂,新鍵未完全形成”微觀狀態的理解是整個化學動力學學習的難點。因此,采用多種手段搜索過渡態,并直觀、清晰地展示出溫度對反應速率常數的影響,對物理化學相關知識點的學習具有重要意義[2,3]。
本文中,我們以1,3-丁二烯與乙烯的環加成反應為例,從微觀角度出發,通過計算化學軟件演示TS、QST2、QST3三種關鍵詞搜索過渡態的過程,向學生直觀地展示異構反應過渡態的立體構型;通過計算不同溫度下的反應速率常數,揭示溫度對反應速率的影響程度,幫助學生深入理解反應機理以及化學反應的本質。
2 實施過程
狄爾斯-阿爾德反應(Diels-Alder reaction)是有機合成中的一類重要反應,通常用于C―C鍵的形成。其中1,3-丁二烯和乙烯的反應是最典型的狄爾斯-阿爾德反應,其方程式如圖1所示。
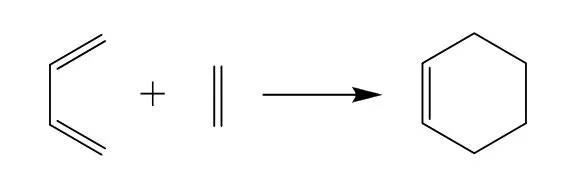
圖1 1,3-丁二烯和乙烯反應制環己烯
本文以1,3-丁二烯與乙烯的環加成反應為例,通過Gaussian16[4]、GaussView6.0[5]等計算化學軟件和Origin等數據處理軟件,在B3LYP[6,7]/6-311+G(d,p)[8,9]水平下,實現多手段的反應過渡態搜索并獲得寬溫度范圍的反應速率常數。具體過程可分為以下四步(圖2):(1) 構建反應物和產物初始結構并進行幾何優化,找到勢能面上的穩定點,得到分子的能量;(2) 使用TS、QST2、QST3三種方法搜索過渡態,并對搜索到的過渡態進行頻率計算和內稟反應坐標(IRC)驗證;(3) 計算反應過程中熱力學函數的變化,構建反應勢能面,展示反應機理;(4) 根據(3)中得到的自由能壘,結合Arrhenius公式,揭示溫度對反應速率常數的影響。
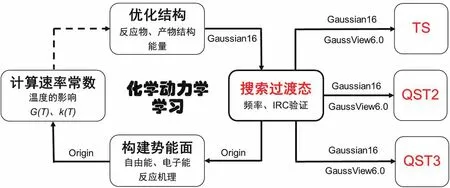
圖2 計算化學方法輔助化學動力學學習流程圖
3 過渡態的搜索與驗證
3.1 反應物、產物分子的結構優化
構建反應物和產物結構并進行幾何優化,尋找到能量局域最低的穩定點(能量對坐標的一階導數為0)是搜索反應過渡態的第一步。我們通過GaussView6.0軟件分別構建反應物(C4H6,C2H4)和產物(C6H10)的初始構型,并對其進行優化。如圖3(a)、(b)所示,反應物分子1,3-丁二烯中的C=C鍵長(1.337 ?)大于乙烯的C=C鍵長(1.329 ?),產物分子環己烯的C=C鍵長(1.334 ?)相比1,3-丁二烯中的略短,但C―C鍵長有較大的差別。構型的改變對C=C以及C―C的鍵長影響較大。
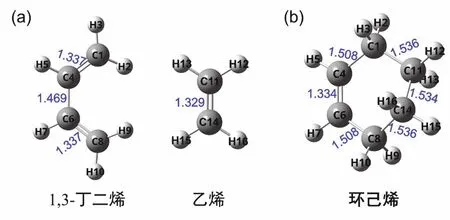
圖3 反應物和產物分子的幾何構型(鍵長單位為?,1 ? = 0.1 nm)
3.2 三種方法搜索過渡態
Gaussian16提供了三種搜索過渡態的方法,分別通過三個關鍵詞來執行:TS (輸入文件中只需要提供過渡態的初猜結構)、QST2 (輸入文件中需要提供反應物和產物結構)、QST3 (輸入文件中需要提供反應物、產物結構和過渡態的初猜結構)。
3.2.1 關鍵詞TS搜索過渡態
(1) 過渡態模型構建。為了能夠順利找到過渡態,首先需要給出一個合理的過渡態結構的初始猜測。如何對過渡態進行初猜?我們可以根據優化出的反應物和產物構型,結合過渡態的特點(舊鍵未完全斷裂,新鍵未完全形成),對過渡態進行合理的初猜。在本例中,反應物分子發生環加成反應轉變成產物主要是乙烯分子的C11和C14原子分別進攻1,3-丁二烯分子的C1和C8原子,同時C1=C4、C6=C8和C11=C14雙鍵斷裂,C4=C6雙鍵形成(原子編號見圖3(a))。因此我們可將過渡態初猜結構設定為C11和C14原子靠近C1和C8原子,并且拉長C1=C4、C6=C8和C11=C14雙鍵,縮短C4―C6單鍵。
(2) 搜索過程。在使用GaussView6.0構建出過渡態的構型后,我們就可以使用Gaussian16軟件中的TS關鍵詞搜索過渡態。具體步驟如下:點擊Calculate → Gaussian Calculation Setup,打開Gaussian的計算設置對話框,① 設置任務類型,選擇Job Type → Opt + Freq (幾何優化和頻率計算),并選擇優化到過渡態(TS(Berny)),選擇Force Constants:Calculate at First Point,并且在下方的Additional Keywords里輸入opt=noeigen,② 設置計算方法和基組等,選擇Method → Method:Ground State、DFT、B3LYP,Basis Set:6-311+G(d,p),Charge:0,Spin:Singlet;③ 設置作業名稱,選擇Title →輸入TS;④ 設置計算的chk文件名稱,選擇Link 0 → Options,設置Chkpoint File → Specify,TS.chk(計算生成的chk文件命名為TS.chk)。⑤ 設置完成后,點擊Submit,保存輸入文件,提交至Gaussian16程序計算。
3.2.2 關鍵詞QST2搜索過渡態
(1) 輸入文件的準備。將優化得到的反應物和產物結構寫入同一個輸入文件中,Gaussian程序會根據我們輸入的反應物和產物結構自動生成一個過渡態的初始結構。打開GaussView6.0,粘貼優化好的反應物分子結構,點擊File → Add to Molecule Group,粘貼優化好的產物分子結構。點擊Tools → Atom List,打開原子列表編輯器,在Tag列修改原子序號,確保反應物和產物分子的原子序號一致。
(2) 搜索過程。點擊Calculate → Gaussian Calculation Setup,打開計算設置對話框,設置任務類型Job Type → Opt + Freq,選擇優化到過渡態(TS(QST2)),選擇Force Constants:Calculate at First Point,并且在下方的Additional Keywords里輸入opt=noeigen。方法和基組等其他設置同TS關鍵詞搜索過渡態相同。設置完成后,點擊Submit,提交至Gaussian16進行計算。
3.2.3 關鍵詞QST3搜索過渡態
(1) 輸入文件的準備。將優化得到的反應物、產物結構以及過渡態的初猜結構寫入同一個輸入文件中,Gaussian程序將根據輸入的這三個構型重新生成一個過渡態的初始猜測結構。打開GaussView6.0,粘貼優化好的反應物分子結構,點擊File → Add to Molecule Group,粘貼優化好的產物分子結構,繼續點擊File → Add to Molecule Group,構建過渡態的初猜結構。點擊Tools → Atom List,打開原子列表編輯器,在Tag列修改原子序號,確保反應物、產物和過渡態分子的原子序號一致。
(2) 搜索過程。點擊Calculate → Gaussian Calculation Setup,打開計算設置對話框,設置任務類型Job Type → Opt + Freq,選擇優化到過渡態(TS(QST3)),選擇Force Constants:Calculate at First Point,并且在下方的Additional Keywords里輸入opt=noeigen。方法和基組等其他設置同TS關鍵詞搜索過渡態相同。設置完成后,點擊Submit,提交至Gaussian16進行計算。
圖4(a)顯示了TS方法優化后的反應過渡態的結構,C11原子進攻C1原子,同時C14原子進攻C8原子,C1、C4、C6、C8、C14與C11原子共同形成了六元環過渡態。將要斷裂的C11=C14鍵長為1.389 ?、C1=C4和C6=C8鍵長為1.384 ?,將要形成的C4=C6鍵長為1.402 ?、C1―C11和C8―C14鍵長為2.246 ?。
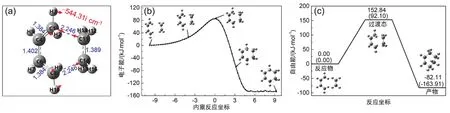
圖4 (a) 過渡態結構及虛頻振動方向(鍵長單位為?);(b) IRC曲線;(c) 環加成反應自由能面(括號內為電子能)
3.2.4 過渡態的確認
(1) 頻率分析。在反應過程中,勢能面上有一最小能量路徑(minimum energy path,MEP)連接著反應物、過渡態和產物的結構。過渡態是最小能量路徑上的能量最高點,也是一階鞍點,它的能量對坐標的一階導數為零,在反應坐標方向上對坐標的二階導數為負,在其他方向上為正。當振動頻率僅有一個虛頻時,就證明這是一個過渡態。使用GaussView6.0軟件查看過渡態的頻率計算結果以及頻率的振動方向可知,振動頻率中只有一個虛頻(544.31icm?1),且虛頻的振動方向對應于C1―C11和C8―C14的生成(圖4(a))。以上分析結果進一步證明了搜索到的結構確實為環加成反應過渡態。
(2) 反應路徑驗證分析。為了驗證該過渡態結構是連接反應物和產物的過渡態,我們進行了內稟反應坐標(IRC)計算。IRC計算從過渡態開始,根據能量降低的方向尋找過渡態連接的兩個極小值,IRC是連接反應物、過渡態和產物的能量最低的反應路徑。計算結果表明,IRC曲線上每個點都對應一個亞穩態結構(如圖4(b)所示),能量最高點對應過渡態結構,曲線兩端分別接近反應物和產物結構,從圖中可以明顯看出環加成反應過程伴隨著C1―C11和C8―C14、C4=C6的生成以及C1=C4、C6=C8、C11=C14的斷裂。
3.3 反應勢能面的構建
反應物、過渡態和產物的電子能、內能、焓和吉布斯自由能值如表1所示。在298.15 K、101.325 kPa下,該環加成反應(C4H6+ C2H4→ C6H10)的ΔrHm= ?145.34 kJ·mol?1,ΔrUm= ?142.86 kJ·mol?1,ΔrGm=?82.11 kJ·mol?1,為熱力學自發過程。圖4(c)顯示了B3LYP/6-311+G(d,p)水平下反應的吉布斯自由能面,反應物分子1,3-丁二烯和乙烯反應生成產物環己烯過程放熱82.11 kJ·mol?1,反應物需要越過152.84 kJ·mol?1的能壘才能轉變成產物。結合《物理化學》教材中的化學動力學原理可知,該反應在常溫下的速率非常緩慢,需要升高溫度才能有效提高環己烯的產率。

表1 B3LYP/6-311+G(d,p)水平下反應物、過渡態和產物的電子能E、內能U、焓H和吉布斯自由能G
3.4 溫度對反應速率常數的影響
結合物理化學中的Arrhenius公式,熱力學形式的反應速率常數的計算公式如下:
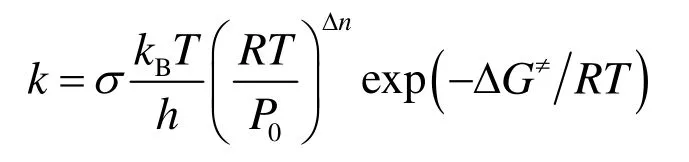
其中σ是反應路徑的簡并度,kB是玻爾茲曼常數,T是反應溫度,h是普朗克常數,R是摩爾氣體常數,P0是標準壓力;Δn = n? 1,n是所有反應物的計量系數之和;ΔG≠是反應的活化吉布斯自由能,即過渡態和反應物的自由能之差。
通過3.3中的計算結果可知,298.15 K時,反應物到產物的活化吉布斯自由能ΔG≠為152.84 kJ·mol?1。因此,298.15 K時反應的速率常數為5.14 × 10?16(mol·m?3)?1·s?1。
此外,化學動力學理論表明升高溫度有利于提升反應速率。那么,溫度對該異構反應速率常數的影響有多大呢?通過計算不同反應溫度下(373.15、423.15、473.15、523.15和573.15 K)的環加成反應速率常數(表2),我們發現隨著反應溫度的升高,自由能壘逐漸升高,且反應速率常數有5個數量級的提升(373.15 K–573.15 K)。
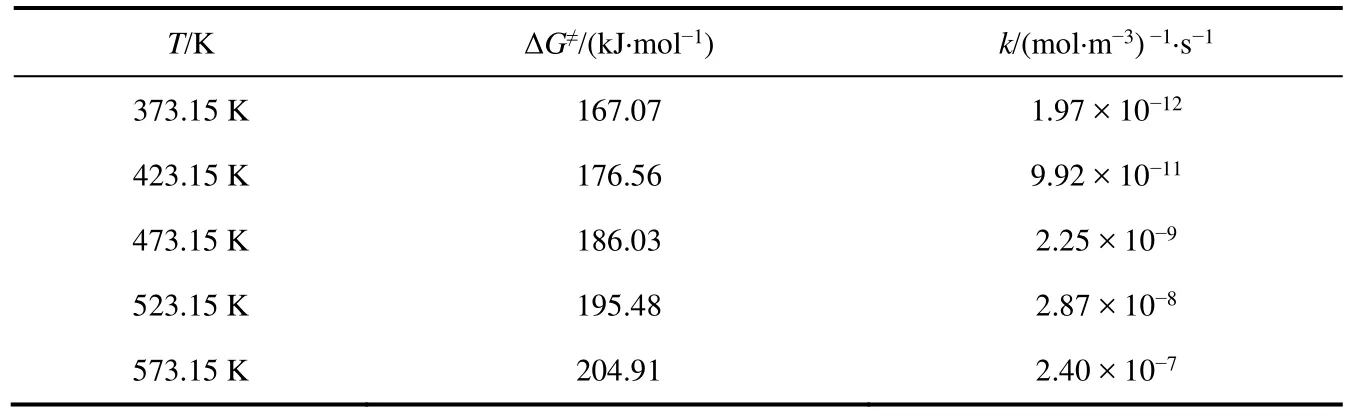
表2 不同溫度下環加成反應的活化吉布斯自由能ΔG≠和反應速率常數k
4 結語
本文以1,3-二丁烯和乙烯的環加成反應中過渡態的搜索為例,介紹了三種搜索反應過渡態的方法及其詳細操作步驟。本文不僅通過計算化學可視化軟件將過渡態這一抽象的概念進行了直觀展示,而且討論了了溫度對反應速率的影響,以期實現幫助學生深入理解化學動力學理論知識、激發對物理化學的學習興趣、培養創新思維能力和科研能力的目的。
猜你喜歡
房地產導刊(2022年5期)2022-06-01 06:20:14
哲學評論(2021年2期)2021-08-22 01:53:34
建材發展導向(2021年12期)2021-07-22 08:06:48
建材發展導向(2021年7期)2021-07-16 07:07:52
中學生數理化(高中版.高二數學)(2021年12期)2021-04-26 07:43:48
中學生數理化(高中版.高考數學)(2021年12期)2021-03-08 01:28:50
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
現代企業(2015年9期)2015-02-28 18:56:50
