硼摻雜對LiNi0.825 Co0.115 Mn0.06 O2高鎳正極材料微觀形貌及電化學性能的影響①
2021-09-15 13:35:00陳嘉鑫李靈均
礦冶工程 2021年4期
關鍵詞:結構
陳嘉鑫,李靈均,譚 磊
(長沙理工大學 材料科學與工程學院,湖南 長沙410114)
鋰離子電池高鎳正極材料具備高能量密度,能夠滿足現有電動交通工具(EV)對于高續航里程的要求,是現在市場中最受青睞的正極材料之一[1-3]。然而,組分中高鎳含量給材料帶來高容量的同時也使材料穩定性變差,儲存過程中極易與空氣組分反應,同時在反復的充放電循環過程中更加容易發生結構衰退,發生結構中晶格氧的釋放,繼而引發熱失控造成嚴重安全問題[4],限制了高鎳材料的進一步應用。為解決上述問題,多采用摻雜改性來提升高鎳材料的穩定性[5]。
在諸多摻雜元素中,硼元素原子半徑小,且化合物熔點較低,在高溫燒結過程中容易固相擴散到高鎳材料的內部形成晶格摻雜。譬如Yang等人在600℃二次熱處理制備了硼濃度梯度修飾的高鎳正極材料,改性后材料性能得到了有效提升[6]。
本文通過一步式固相燒結法,合成了硼摻雜修飾的高鎳正極材料,以期提升材料的穩定性和電化學性能。
1 實 驗
1.1 實驗原料及材料合成方法
高溫固相法一步式合成硼摻雜高鎳LiNi0.825Co0.115-Mn0.06O2正極材料:將H3BO3(99%,國藥集團)、LiOH(98%,西隴化工)和Ni0.825Co0.115Mn0.06(OH)2前驅體(長遠鋰科)按比例在研缽中研磨混合均勻(LiOH∶(前驅體+H3BO3)=1.05∶1)。硼含量分別為前驅體摩爾數的0%(編號NCM)、1%(編號B1)、2%(編號B2)和3%(編號B3)。然后將混合原料置于管式爐中,在氧氣氣氛條件下先480℃預燒5 h,再750℃煅燒12 h,自然冷卻后取出。
1.2 結構形貌表征和電化學性能測試
使用X射線衍射儀(Bruker D8 Advance,Germany)對材料進行物質晶相結構組成分析,通過掃描電子顯微鏡(TESCAN MIRA3,Czech)分析材料的微觀形貌結構。
按照質量分數80%、10%、10%將活性正極材料、活性炭黑、PVDF加NMP混合涂布到鋁箔上,烘干后制備得到極片;使用電池級金屬鋰片為負極;以1 mol/L的LiPF6溶解于EMC∶DMC∶EC(體積比)=1∶1∶1的混合有機溶液(外加1%VC為添加劑)為電解液;隔膜為玻璃纖維Celgard2400;在高純氬氣氣氛手套箱中組裝為扣式電池,進行電化學測試。
2 實驗結果及討論
2.1 XRD物相表征
不同含量硼摻雜高鎳正極材料XRD圖如圖1所示。由圖1可知,原始樣和所有硼摻雜高鎳正極材料的XRD圖譜中未見雜質峰,都呈現出標準的α-NaFeO2層狀相(R-3m空間群)。同時,(006)/(012)和(018)/(110)位置具有良好的劈裂,意味著各個材料都保持著良好的層狀結構。同時,經過硼摻雜后樣品的(003)峰向著低角度偏移,可知層間距變大。此外,所有樣品的I003/I104值均在1.6以上(見表1),說明材料結構中陽離子混排較低[7]。但是隨著硼的占比提高,I003/I104峰強比值下降,可能是硼的添加加劇了材料結構中的陽離子混排[6]。此外,材料的晶格常數a和c都隨之增大,說明硼的添加導致了材料結構晶格的膨脹。由于B離子的半徑為0.027 nm,小于Li和過渡金屬Ni、Co、Mn的半徑,所以B是摻雜進入晶體結構間隙而非取代原有元素位置[8]。
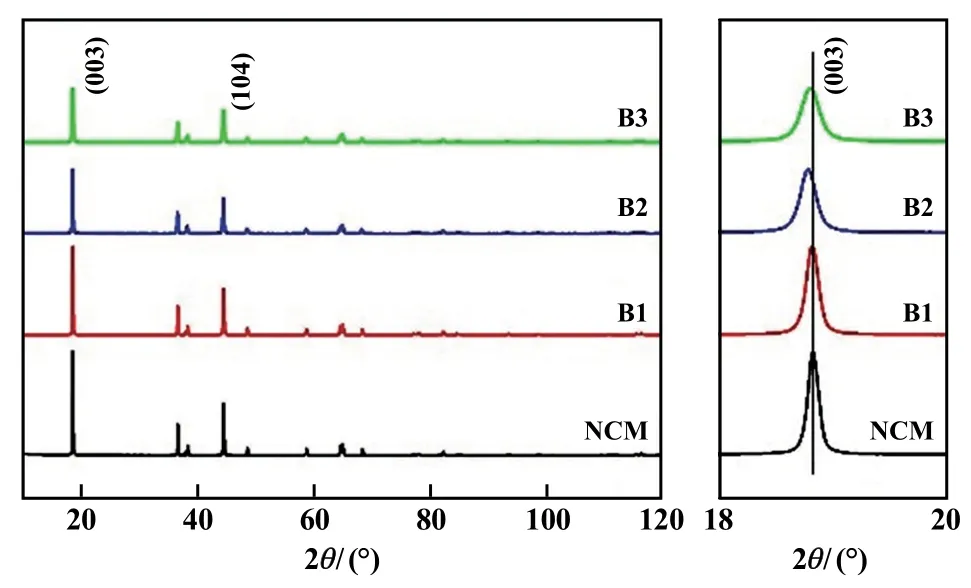
圖1 不同含量硼摻雜高鎳正極材料XRD圖及(003)峰局部放大圖
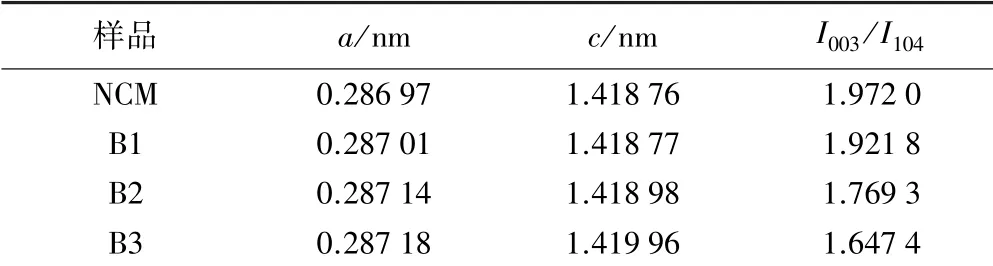
表1 不同含量硼摻雜高鎳正極材料的晶胞參數
2.2 微觀形貌結構表征
不同含量硼摻雜高鎳正極材料的SEM圖見圖2。各個材料的二次顆粒均為粒徑10~20μm的球狀顆粒,由一次顆粒組裝團聚組成。隨著硼的添加,一次顆粒形貌由原始樣品不規則排列的類橢球狀(粒徑約200~300 nm)逐漸變為細長紡錘狀(長約700~800 nm),說明硼摻雜能夠起到調節材料微觀形貌的作用。

圖2 不同含量硼摻雜高鎳正極材料SEM圖
不同含量硼摻雜高鎳正極材料的截面SEM圖見圖3。從圖3可知,NCM材料中一次顆粒相互之間的間隔明顯,排列無序;而B2材料一次顆粒之間輪廓變模糊,成徑向排列。結合EDS-mapping圖可知,B均勻分散到了材料內部,可認為硼的引入改變了材料的晶面形成能,織構了這種徑向排列的微觀形貌結構[9]。

圖3 不同含量硼摻雜高鎳正極材料的截面SEM圖
通過BET方法檢測了材料的比表面積,結果見表2。經過硼修飾的材料比表面積相對NCM出現了明顯增長,可能會加劇材料和電解質之間的界面反應。

表2 材料比表面積
2.3 硼摻雜LiNi0.825 Co0.115 Mn0.06 O2正極材料電化學性能測試
不同含量硼摻雜高鎳正極材料電化學性能見圖4。由圖4可見,在0.1C倍率、2.7~4.3 V范圍內進行充放電測試,材料的首次充放電效率隨硼摻雜量增加而降低,NCM首次放電比容量為200.10 mAh/g;而B3樣品首次放電比容量下降到了189.44 mAh/g。主要原因是硼的引入導致了結構中陽離子混排的加劇,從而造成可逆容量下降[6]。此外,經過硼摻雜后,材料首次充電曲線的初始電壓平臺略微升高,可歸因于摻雜的硼元素不具備電化學活性,導致材料內部阻抗增加、極化增大。
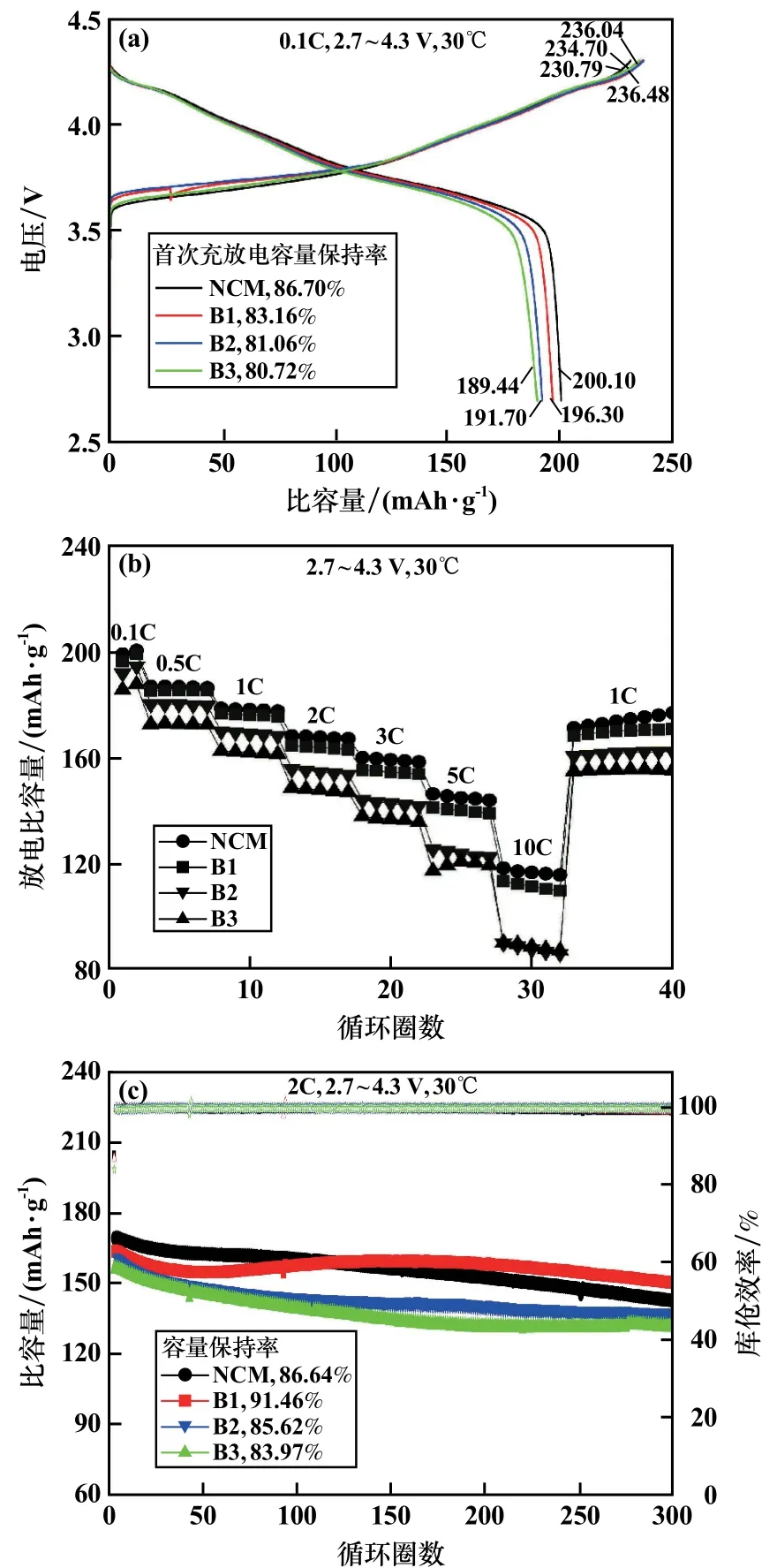
圖4 不同含量硼摻雜高鎳正極材料電化學性能圖
在倍率性能方面,相比于NCM樣品,B1材料的倍率性能略微下降,B2和B3的倍率性能更差,特別是在10C的高倍率下比容量僅80 mAh/g,這是由于摻雜的硼元素在結構中為固定的+3價,不具有電化學活性,降低了材料的電子電導造成的[9]。
在2C電流密度下,2.7~4.3 V電壓范圍循環300圈后,B1容量保持率高達91.46%,比原始樣品的86.64%高出了將近5個百分點,這是由于B元素容易進入材料晶格結構內部與氧鍵合成B—O鍵,提升了結構的穩定性。此外,經過硼摻雜后的材料為一次顆粒徑向排列的結構,有助于緩解材料工作過程中各向異性的體積相變所造成的應力堆積,從而提升材料的循環性能[10]。B2和B3材料的循環性能下降可能是由于材料結構中存在過多不具有電化學活性的硼元素,占據了四面體間隙位置,抑制鋰離子的擴散,增加了材料的阻抗[11]。
不同含量硼摻雜高鎳正極材料循環伏安(CV)曲線見圖5。在掃描速度0.1 mV/s、掃描電壓2.7~4.5 V范圍內,材料前3圈的CV曲線中首圈循環的氧化峰電位更高,第2和第3圈的曲線高度重疊,這對應了首次充放電過程中的不可逆容量損失,與固體電解質膜(SEI膜)的形成有關[12]。經過硼摻雜過后材料的氧化還原電位差值ΔV分別為0.221 V、0.241 V和0.191 V,都高于未摻雜樣品的0.163 V,說明硼摻雜增大了材料的極化。特別是B3樣品,第2圈和第3圈電位差值相較于首圈值繼續增大,意味過量硼摻雜會降低材料電極反應的可逆性。

圖5 不同含量硼摻雜高鎳正極材料的循環伏安曲線
對NCM和B1材料進行了循環前后的EIS表征,在4.3 V充電態進行測試,以驗證硼摻雜對材料循環前后鋰離子擴散動力學的影響,結果見圖6。循環前NCM的表面膜阻抗(Rf)和電荷轉移阻抗(Rct)分別為39.47Ω和52.38Ω,而B1的Rf和Rct分別為40.25Ω和69.32Ω。B1比NCM的初始Rct更低歸因于硼的摻雜降低了材料的電導率。在2C下300圈循環后,NCM的Rf和Rct分別為44.3Ω和347.6Ω,而B1的Rf和Rct分別為49.55Ω和168.8Ω,NCM的Rct增量更大,說明其經過循環后材料發生的結構衰退更為嚴重。此外擬合計算了300圈充放電循環后的鋰離子擴散系數,NCM和B1的值分別為2.78×10-13cm2/s和6.88×10-13cm2/s,證實了B1材料經過循環后仍然保持更好的鋰離子擴散能力。
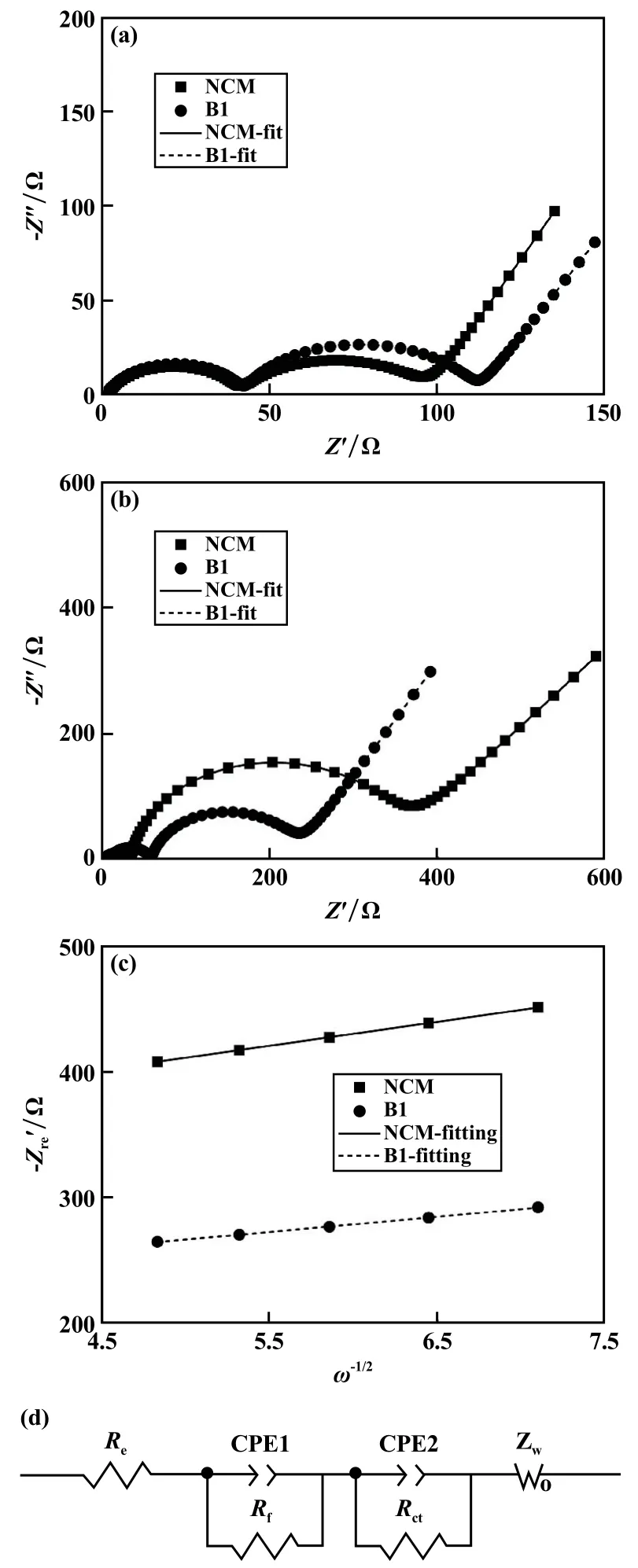
圖6 NCM和B1的EIS圖譜
3 結 論
1)高鎳正極材料經硼摻雜改性后,結構中的Ni/Li陽離子混排升高、晶格膨脹、比表面積增大;一次顆粒形貌由類橢球狀變為中心放射狀排列的紡錘型,有助于緩解循環過程中的應力堆積問題。
2)經過1%硼摻雜的正極材料B1循環性能相對于原始材料NCM有著顯著提升,在2.7~4.3 V電壓區間進行2C倍率充放電循環300圈后,容量保持率仍有91.46%。這歸因于B摻雜到材料晶格間隙中并與O形成B—O鍵,提升了材料的結構穩定性;另外B1具有中心放射狀排列的細長紡錘結構,有助緩解循環中的應力堆積,優化了材料循環性能。
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
