高效液相色譜法測定口服液體制劑中12種添加劑的含量
2021-09-01 03:13:54辜慧李道霞何成軍杜鋼四川省食品檢驗研究院成都611731
中南藥學 2021年7期
關鍵詞:檢測
辜慧,李道霞,何成軍,杜鋼(四川省食品檢驗研究院,成都 611731)
口服液體制劑易被微生物污染而發霉變質,為保障該類制劑臨床用藥安全,需添加適量的防腐劑,抑制微生物的生長繁殖,達到有效的防腐目的。另外,還需添加適量甜味劑及色素,改善制劑口感及外觀,提高患者依從性[1]。然而,添加劑的超范圍使用、過量添加會對人體造成一定的傷害。人工合成色素、甜味劑和防腐劑在市場上已有濫用的趨勢,給藥品安全帶來了隱患,對消費者的健康造成了直接的威脅。目前,國內口服液體制劑生產企業眾多,所加添加劑的種類與濃度亦各不相同,關于添加劑的測定方法文獻均有報道[2-7],未見有同時測定人工合成色素、甜味劑和防腐劑的方法報道。為了提高檢驗效率,節約成本,更全面地控制口服液體制劑的安全性,本研究建立了一種同時測定口服液體制劑中12種添加劑的高效液相色譜(HPLC)法。
1 儀器與試藥
LC-20AT 型高效液相色譜儀,配二極管陣列檢測器(日本SHIMADZU 公司);ME204 分析天平(十萬分之一,瑞士METTLER TOLEDO 公司);FB15065 超聲波發生器(美國Fisher 公司);Milli-Q 型高純水發生器(美國Millipore 公司);苯甲酸(批號:B0001649,濃度:1000 μg·mL-1)、山梨酸(批號:B0002741,濃度:1000 μg·mL-1)、酸性紅(批號:C0005676,純度:90.7%)、脫氫乙酸(批號:B0003452,純度:99.9%)、糖精鈉(批號:B0001955,濃度:1000 μg·mL-1)(BePure);檸檬黃、日落黃、莧菜紅、胭脂紅、新紅、誘惑紅及亮藍(A CHEMTEK INC,批號:S027451,濃度:1000 μg·mL-1);甲醇、乙酸銨(色譜純,美國Fisher 公司);水為超純水。
2 方法與結果
2.1 色譜條件
色譜柱:Waters XBridge C18(250 mm×4.6 mm,5 μm);流速:0.8 mL·min-1;柱溫:35℃;檢測波長:230 nm;進樣體積:10 μL;流動相A 為20 mmol·L-1乙酸銨溶液,流動相B 為甲醇,梯度洗脫程序見表1。
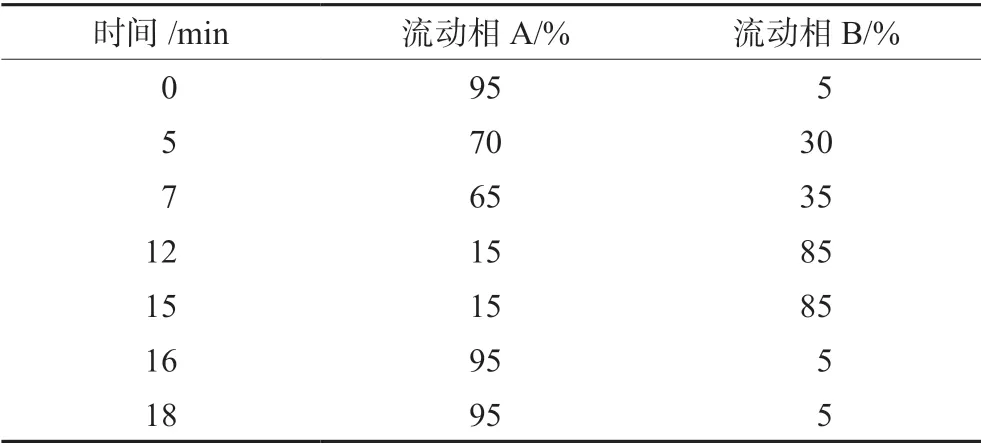
表1 梯度洗脫程序表Tab 1 Gradient elution program
2.2 溶液的配制
2.2.1 混合對照品溶液 分別精密稱取酸性紅、脫氫乙酸對照品約10 mg,分別置10 mL 量瓶中,用水超聲溶解并定容至刻度,搖勻,分別作為酸性紅對照品儲備液、脫氫乙酸對照品儲備液。其余組份對照品儲備液均從對照品公司購得。再分別精密量取各對照品儲備液1 mL,置同一50 mL 量瓶中,加水稀釋至刻度,搖勻,作為混合對照品溶液。
2.2.2 供試品溶液 以布洛芬混懸液為例,精密量取樣品1 mL,置10 mL 量瓶中,加水適量,超聲處理10 min,再加水定容至刻度,搖勻,經0.45 μm 濾膜過濾,取續濾液作為供試品溶液。
2.2.3 陰性樣品溶液 取陰性樣品(蛋白酶合劑)按“2.2.2”項下方法制備陰性樣品溶液,備用。
2.3 方法學考察
2.3.1 專屬性考察 分別量取“2.2”項下的空白溶劑(水)、混合對照品溶液、陰性樣品溶液、供試品溶液,按“2.1”項下色譜條件進樣10 μL,記錄色譜圖,見圖1。結果色譜圖基線穩定,峰形對稱,分離度較好,各峰保留時間適中,能夠滿足檢測需要,說明本試驗的專屬性良好。
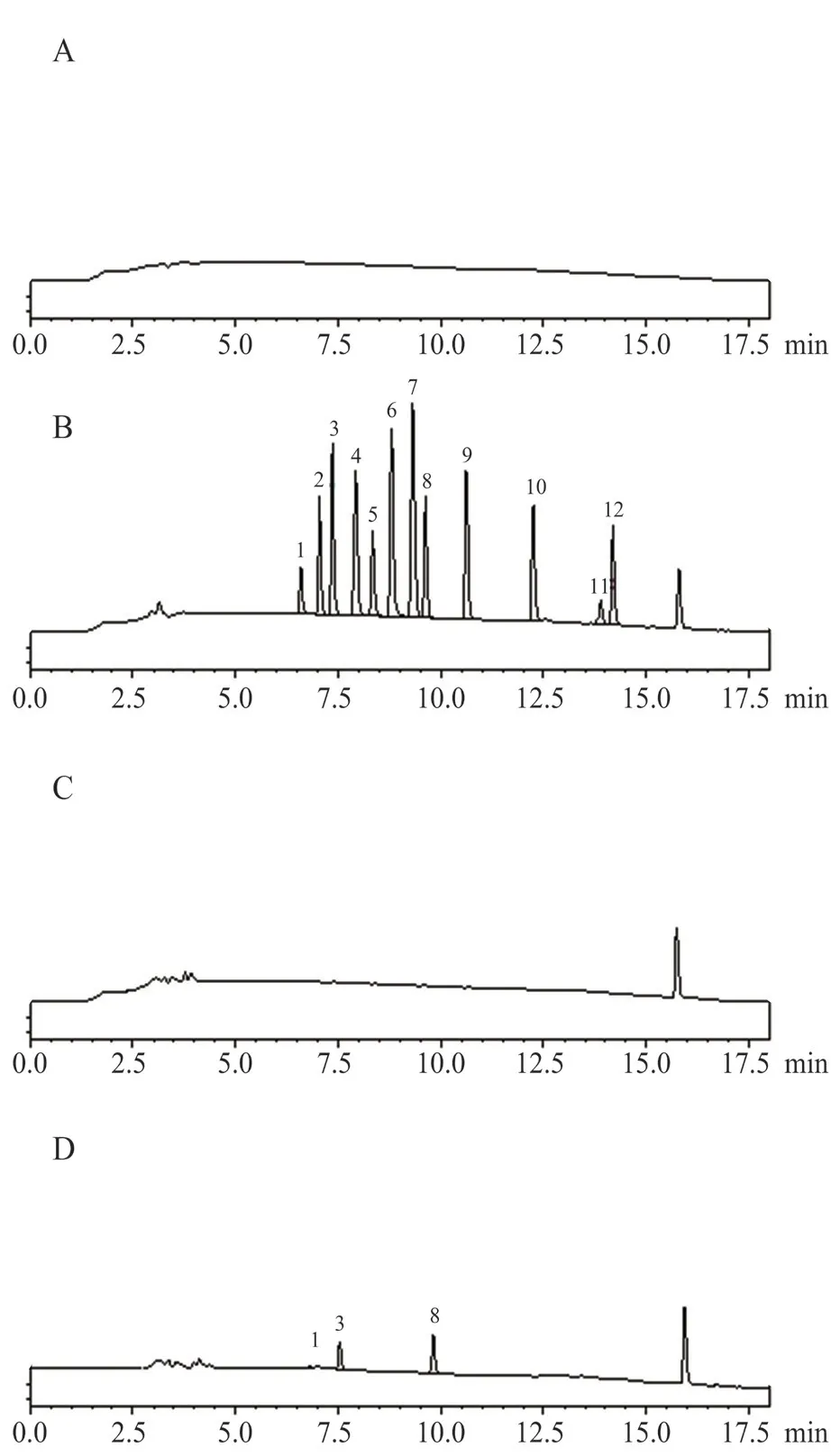
圖1 典型HPLC 色譜圖Fig 1 Typical chromatogram by HPLC
2.3.2 線性關系考察 分別精密量取不同體積的各對照品儲備液,加水稀釋成質量濃度分別為0.5、1、2、5、10、20 μg·mL-1的混合對照品溶液,精密吸取10 μL 注入液相色譜儀,以各組分的峰面積Y為縱坐標,進樣質量濃度X(μg·mL-1)為橫坐標,繪制標準曲線,結果見表2。
2.3.3 檢測限及定量限考察 取混合對照品溶液,逐級稀釋成系列質量濃度,分別進樣10 μL,當峰高為基線噪音的3 和10 倍量時,測得各添加劑的檢測限及定量限,結果見表2。
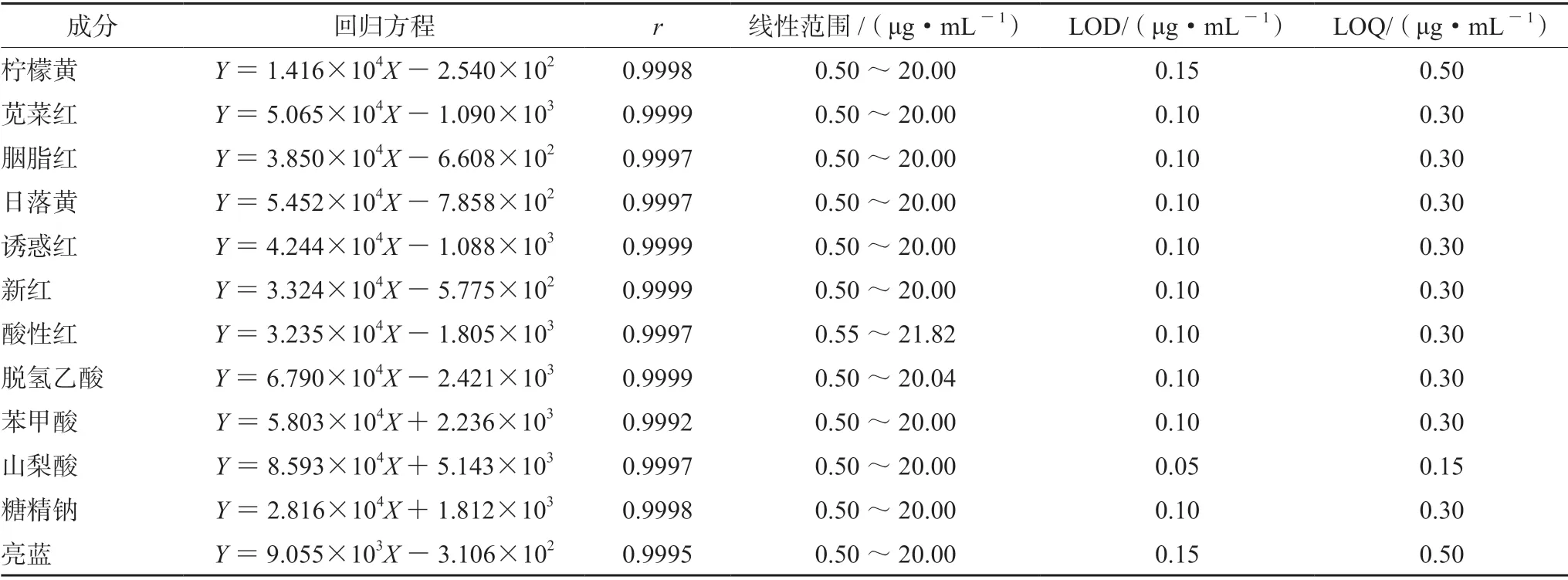
表2 12 種添加劑的回歸方程、相關系數、線性范圍、檢測限與定量限Tab 2 Regression equation,correlation coefficient,linearity,LOD and LOQ for 12 additives
2.3.4 精密度考察 取混合對照品溶液,按“2.1”項下色譜條件下連續進樣6 次,苯甲酸等12 種添加劑峰面積的RSD為0.11%~0.85%(n=6),表明儀器精密度良好。
2.3.5 重復性考察 取一批樣品(檢出檸檬黃、莧菜紅、胭脂紅),按“2.2.2”項下方法前處理,平行制備6 份,按“2.1”項下色譜條件檢測,計算得樣品中檸檬黃平均含量為0.52 μg·mL-1,莧菜紅平均含量為3.11 μg·mL-1,胭脂紅平均含量為5.88 μg·mL-1,RSD分別為1.8%、1.4%、0.99%(n=6),表明方法重復性良好。
2.3.6 回收試驗 以未檢出添加劑的陰性樣品為基質,按低、中、高濃度分別精密加入混合對照品溶液(質量濃度為20 μg·mL-1)0.5、1.0、5.0 mL,按“2.2.2”項下方法處理,制備約相當于1、2、5 μg·mL-1的回收試驗溶液,每個濃度平行制備6 份。按“2.1”項下色譜條件下進行檢測,并計算回收率及RSD,結果見表3。
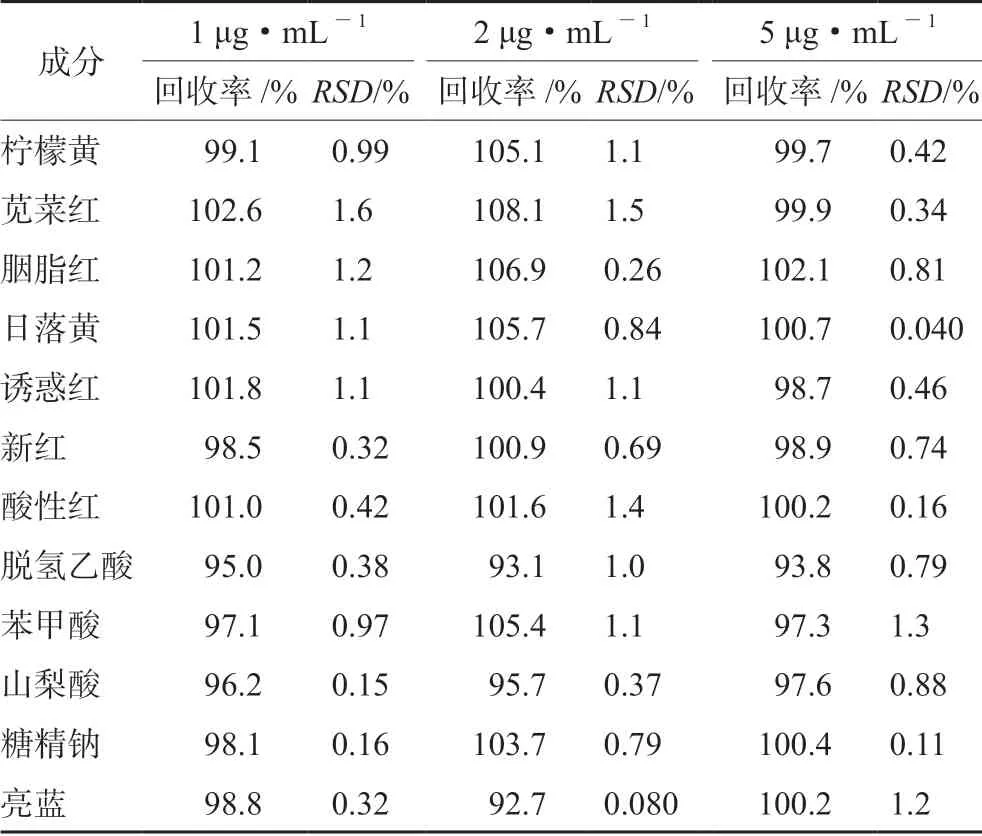
表3 12 種添加劑的平均回收率(n=6)Tab 3 Average recovery of 12 additives (n=6)
2.3.7 穩定性試驗 取回收試驗中的低、中、高濃度的溶液,室溫放置,分別于0、6、12、24、48 h 進樣,各組分峰面積的RSD均小于2.0%,表明12 種添加劑在48 h 內均穩定。
2.3.8 樣品測定 取10 批抽檢的口服液體制劑,按“2.2.2”項下方法制備供試品溶液,再按“2.1”項下色譜條件測定,結果見表4。
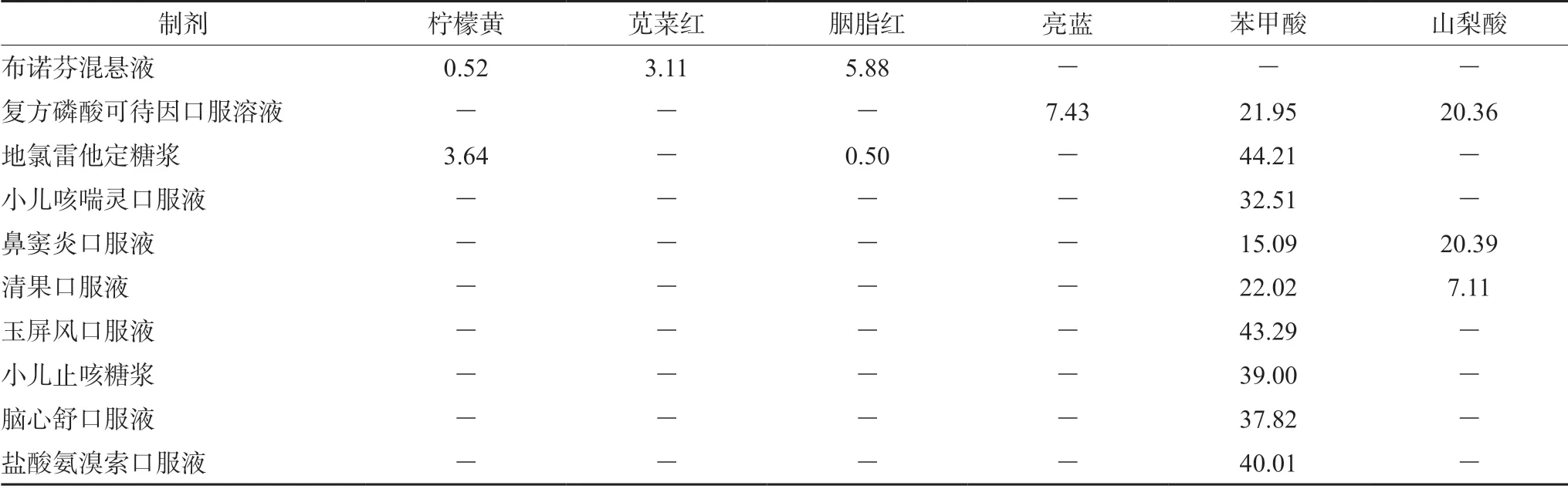
表4 口服液體制劑中檢測出的各成分含量測定結果(μg·mL-1,n=3)Tab 4 Content determination of various constituent in oral liquid preparations (μg·mL-1,n=3)
3 討論
3.1 流動相的選擇
液相色譜方法測定目標化合物時,多在流動相中加入緩沖鹽,試驗通過優化流動相梯度洗脫程序獲得合適的保留時間、峰形以及分離效果。本試驗參考文獻[8-10],流動相選擇乙酸銨溶液-甲醇系統,考察了流動相中乙酸銨的質量濃度分別為10、20、50 mmol·L-1時對12 種添加劑色譜行為的影響。結果表明乙酸銨質量濃度為20 mmol·L-1時,12 種添加劑的色譜峰峰形尖銳,分離度較好。故最終選擇流動相為20 mmol·L-1乙酸銨溶液-甲醇體系,該流動相體系也可在檢出添加劑后,直接用于LC-MS/MS 作進一步分析確認。
3.2 檢測波長的確認
利用二極管陣列檢測器對苯甲酸、山梨酸、糖精鈉、脫氫乙酸、檸檬黃、日落黃、莧菜紅、胭脂紅、新紅、酸性紅、誘惑紅及亮藍分別進行波長掃描。其中苯甲酸、山梨酸、糖精鈉、脫氫乙酸最大吸收波長均在紫外光區,檸檬黃、日落黃、莧菜紅、胭脂紅、新紅、酸性紅、誘惑紅及亮藍雖然最大吸收波長均在可見光區,但紫外光區也有較強吸收。綜合分析12 個標準物質的吸收曲線,各組分在 220~240 nm 均有較強吸收,故選擇230 nm 作為檢測波長,在此波長下基線平穩、噪聲小,各組分均具有較高靈敏度。
3.3 色譜柱的選擇
本文參考文獻[10-12],考察了色譜柱Waters XBridge C18(250 mm×4.6 mm,5 μm)、Agilent ZORBAX SB-C18(4.6 mm×250 mm,5 μm)、Eclipse XDB-C18(4.6 mm×250 mm,5 μm)對目標化合物分離度及峰形的影響,最終選擇 Waters XBridge C18色譜柱,結果12 個目標化合物可以達到分離測定的要求,且準確可靠、重復性好。
3.4 其他
本試驗采用HPLC 法同時測定口服液體制劑中的苯甲酸、山梨酸、糖精鈉、脫氫乙酸、檸檬黃、日落黃、莧菜紅、胭脂紅、新紅、酸性紅、誘惑紅及亮藍的含量,該方法操作簡便、精密度好、回收率高,能滿足檢測要求,適用于口服液體制劑中人工合成色素、甜味劑和防腐劑的檢查。結果發現目前國內一些藥品中仍存在著添加劑濫用的現象,存在較大的安全隱患,需要進行嚴格監管。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
