基于高效液相色譜特征圖譜和一測多評法的淫羊藿配方顆粒質量標準研究
2021-09-01 03:13:48劉曉霞丁青陳芳梁月儀魏梅孫冬梅田維忠霍文杰李振雨廣東一方制藥有限公司廣東佛山528244廣東省中藥配方顆粒企業重點實驗室廣東佛山528244中國中藥控股有限公司廣東佛山5280
中南藥學 2021年7期
關鍵詞:特征
劉曉霞,丁青,陳芳,梁月儀,魏梅,孫冬梅,田維忠,霍文杰,李振雨*(1.廣東一方制藥有限公司,廣東 佛山 528244;2.廣東省中藥配方顆粒企業重點實驗室,廣東 佛山 528244;.中國中藥控股有限公司,廣東 佛山 5280)
中藥配方顆粒作為臨床飲片的補充,具有安全、衛生、便于隨身攜帶、方便調劑使用等優勢,是中藥飲片劑型改革的主要方向[1]。截止目前中藥配方顆粒尚沒有正式出臺統一的國家標準,因此,市場流通的中藥配方顆粒規格不統一,質量的參差不齊導致臨床使用療效難以保證,也為市場監管帶來一定難度。
淫羊藿為小檗科植物淫羊藿Epimedium brevicornuMaxim.、箭葉淫羊藿Epimedium sagittatum(Sieb.et Zucc.)Maxim.、柔毛淫羊藿Epimedium pubescensMaxim.或朝鮮淫羊藿Epimedium koreanumNakai 的干燥葉[2],是臨床常用的補陽藥。現代化學和藥理學研究表明,淫羊藿含有黃酮、酚苷、多糖和生物堿等多種有效成分,其中代表性的有效成分有淫羊藿苷,朝藿定A、B、C,寶藿苷Ⅰ等[3],具有一定的骨細胞誘導潛能[4],能夠促進成骨細胞和骨髓基質細胞的增殖[5],抑制腫瘤細胞表達、阻止骨細胞鈣化的功能[6]。由于中藥有效成分的多樣性及復雜性,單一成分定量的化學藥品質量控制模式既缺乏專屬性,又難以反映中藥內在質量,中藥指紋/特征圖譜能夠提供更為全面、豐富的信息,因此常用于中藥的質量控制研究。一測多評模式借助相對校正因子,只需測定一個成分(內參物),實現對多個有效成分的定量分析,解決了對照品難以獲得的難題,在中藥的質量控制方面應用越來越廣泛[7-9]。本研究將淫羊藿配方顆粒特征圖譜與一測多評模式相結合,從定性定量兩個方面來評價淫羊藿配方顆粒的質量,為淫羊藿配方顆粒質量標準的制訂提供參考。
1 儀器與試藥
1.1 儀器
Waters 高效液相色譜儀(e2695,沃特世公司),Thermo 高效液相色譜儀(U3000,賽默飛公司),Agilent 高效液相色譜儀(1260,安捷倫公司),Agilent ZORBAX SB C18(4.6 mm×250 mm,5 μm)色譜柱(安捷倫公司),Phenomenex Luna C18(4.6 mm×250 mm,5 μm)色譜柱(賽默飛公司),Waters XSelect HSS T3(4.6 mm×250 mm,5 μm)色譜柱(沃特世公司),萬分之一分析電子天平(ME204E)、百萬分之一分析電子天平(XP26,梅特勒-托利多公司),電子天平(JJ600,常熟市雙杰測試儀器廠),數控超聲波清洗器(KQ500DE,昆山市超聲儀器有限公司),恒溫水浴鍋(HWS28型,上海一恒科技有限公司),超純水系統(Milli-Q Direct,默克股份有限公司)。
1.2 試藥
乙腈(默克股份有限公司,色譜純),水為超純水(實驗室自制),其余試劑皆為分析純。朝藿定A 對照品(批號:150520,含量:99.4%)、朝藿定B 對照品(批號:150518,含量:99.24%)(成都普菲德生物股份有限公司);朝藿定C 對照品(批號:111780-201503,含量:100%)、淫羊藿苷對照品(批號:110737-201516,含量:94.2%)、寶藿苷Ⅰ對照品(批號:111852-201603,含量:99.9%)(中國食品藥品檢定研究院);12 批淫羊藿配方顆粒[廣東一方制藥有限公司生產,批號:YYH01~YYH12,規格:0.5 g/袋(相當于常規飲片10 g)]。
2 方法與結果
2.1 色譜條件
采用Agilent ZORBAX SB C18(4.6 mm×250 mm,5 μm)色譜柱;以乙腈為流動相A,水為流動相B,梯度洗脫(0~30 min,24%~26%A;30~31 min,26%~45%A;31~45 min,45%~47%A);流速為1.0 mL·min-1;柱溫為30℃;檢測波長為270 nm;進樣量為10 μL。
2.2 對照品溶液的制備
精密稱定朝藿定A、B、C 對照品和淫羊藿苷對照品適量,加甲醇制成每1 mL 含朝藿定A 17.201 μg、朝藿定B 36.938 μg、朝藿定C 48.270 μg、淫羊藿苷99.890 μg 的混合對照品溶液,即得。
2.3 供試品溶液的制備
取淫羊藿配方顆粒樣品適量,研細,取約0.25 g,精密稱定,置具塞錐形瓶中,精密加入75%乙醇50 mL,稱定重量,超聲處理(功率250 W,頻率40 kHz)45 min,放冷,再稱定重量,用75%乙醇補足減失的重量,搖勻,濾過,取續濾液,即得。
2.4 特征圖譜方法學考察
2.4.1 精密度試驗 取淫羊藿配方顆粒(批號:YYH11)供試品溶液,按“2.1”項下色譜條件連續進樣6 次,以淫羊藿苷色譜峰為參照峰S,計算其余特征峰的相對保留時間RSD值均不超過0.7%,相對峰面積RSD值均不超過0.11%,說明儀器精密度良好。
2.4.2 重復性試驗 取同一批淫羊藿配方顆粒(批號:YYH11),按“2.3”項下方法制備6 份供試品溶液,按“2.1”項下色譜條件測定,以淫羊藿苷色譜峰為參照峰S,計算其余特征峰的相對保留時間RSD值均不超過0.12%,相對峰面積RSD值均不超過0.14%,表明該方法重復性良好。
2.4.3 穩定性試驗 取淫羊藿配方顆粒(批號:YYH11)供試品溶液,按“2.1”項下色譜條件分別在0、3、6、14、19 和24 h 進樣測定,以淫羊藿苷峰為參照峰S,計算其余特征峰的相對保留時間RSD值均不超過0.18%,相對峰面積RSD值均不超過0.29%,表明供試品溶液在24 h 內穩定性良好。
2.5 淫羊藿配方顆粒特征圖譜的建立
取12 批淫羊藿配方顆粒供試品溶液(批號:YYH01~YYH12),按“2.1”項下色譜條件進樣測定,記錄各批次樣品的特征圖譜,并導出CDF格式,將12 批淫羊藿配方顆粒特征圖譜CDF 格式導入到“中藥色譜指紋圖譜相似度評價系統(2012 版)”,以YYH01 的特征圖譜為參照,對12 批淫羊藿配方顆粒特征圖譜進行共有峰標識,并進行保留時間校正和峰匹配(見圖1),選擇出峰穩定,峰形和分離度較好的7 個共有峰作為淫羊藿配方顆粒的特征峰,建立淫羊藿配方顆粒特征圖譜共有模式(見圖2),參考相關文獻[10],通過對照品的保留時間比對并結合3D 光譜分析確定峰2 為朝藿定A,峰3 為朝藿定B,峰4 為朝藿定C,峰5 為淫羊藿苷,峰7 為寶藿苷Ⅰ,見圖3。以淫羊藿配方顆粒特征圖譜共有模式為參照,計算各樣品特征圖譜的相似度均大于0.9(見表1),說明12 批淫羊藿配方顆粒特征圖譜的整體相似度較高,質量較穩定。
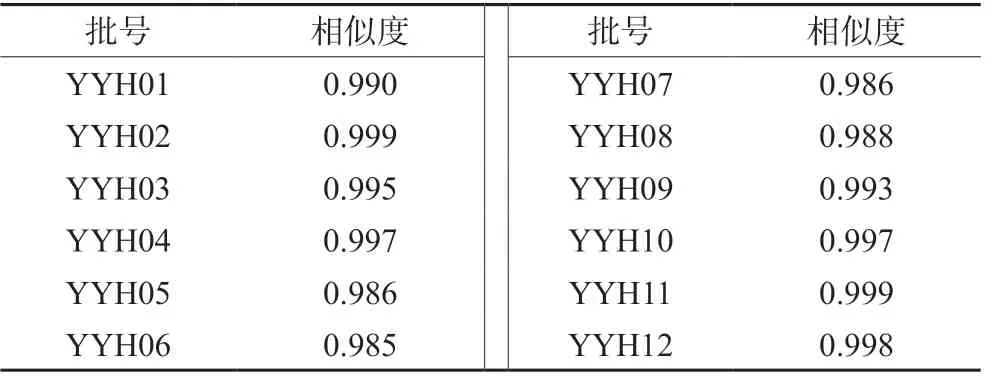
表1 12 批淫羊藿配方顆粒特征圖譜相似度Tab 1 Similarity of specific chromatogram of 12 batches of Herba epimedii dispensing granules
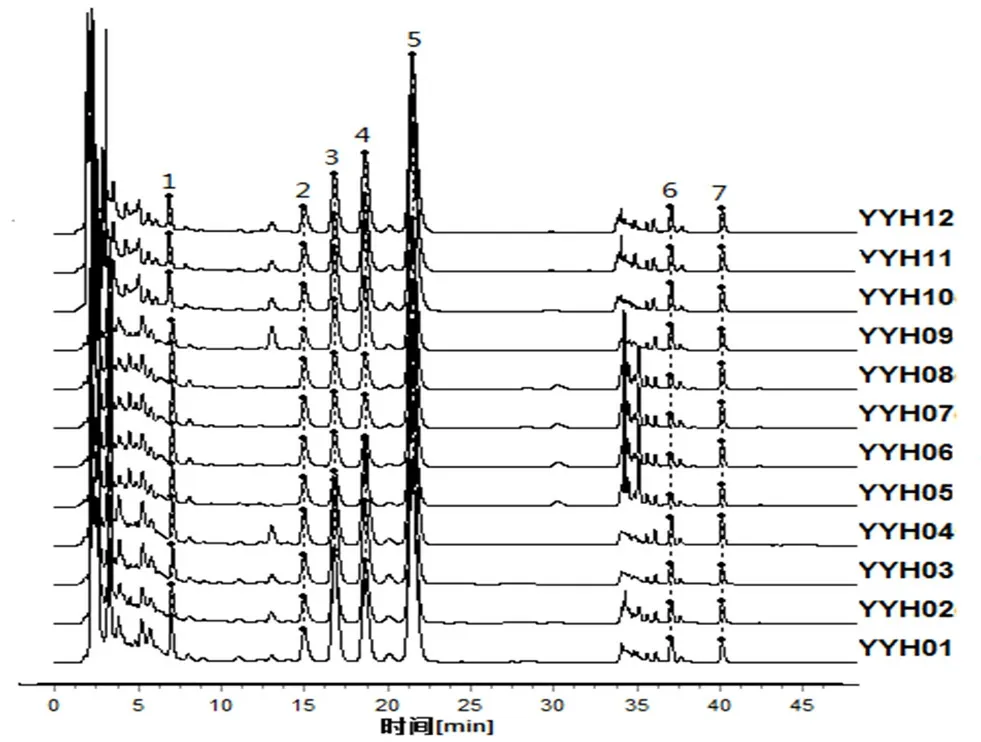
圖1 12 批淫羊藿配方顆粒HPLC 特征圖譜Fig 1 HPLC specific chromatogram of 12 batches of Herba epimedii dispensing granules
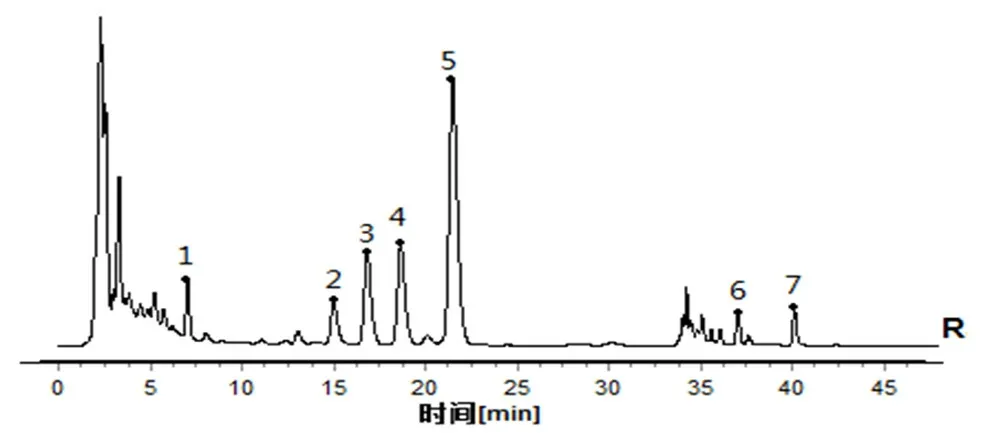
圖2 淫羊藿配方顆粒HPLC 特征圖譜共有模式Fig 2 Common pattern of HPLC specific chromatograms of Herba epimedii dispensing granules
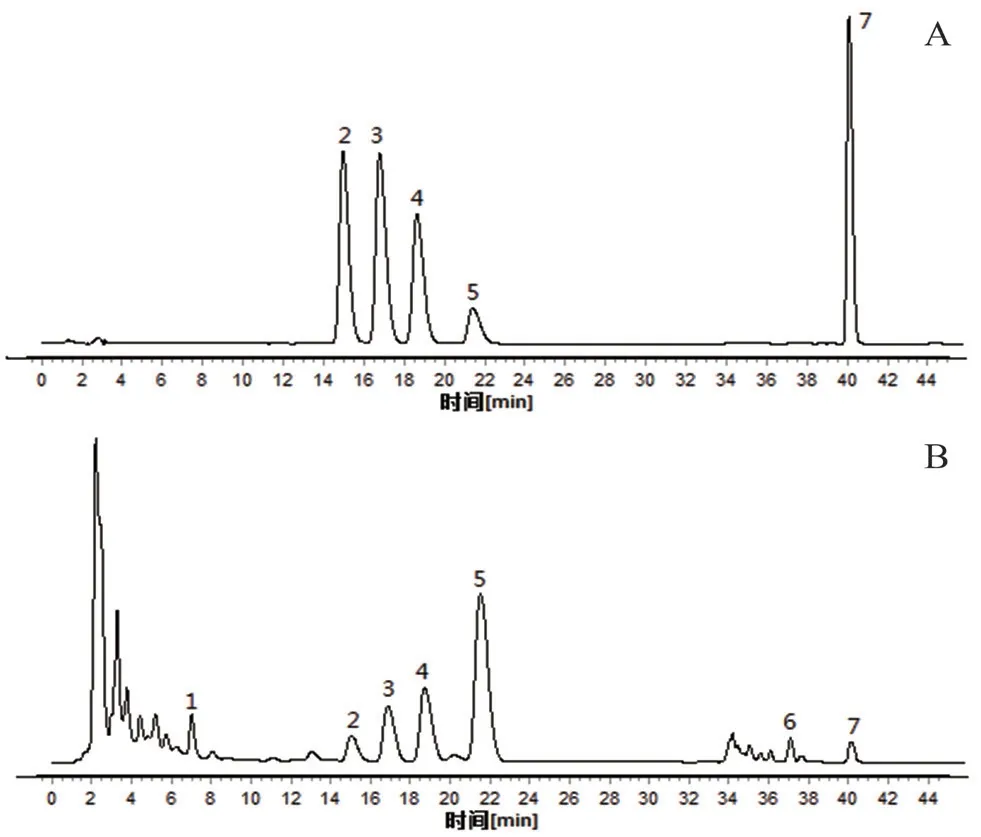
圖3 淫羊藿配方顆粒特征圖譜Fig 3 Chromatogram of Herba epimedii dispensing granules
2.6 總黃酮醇苷一測多評含量測定
2.6.1 線性關系考察 精密稱定對照品朝藿定A 4.883 mg、朝藿定B 4.643 mg、朝藿定C 10.001 mg、淫羊藿苷10.573 mg,置10 mL 量瓶中,加甲醇溶解并定容,制成每1 mL 含朝藿定A 485.370 μg、朝藿定B 460.771 μg、朝藿定C 1000.100 μg、淫羊藿苷995.977 μg 的混合對照品儲備液。精密移取上述混合對照品儲備液0.5、0.5、0.5、1、2、3 mL,分別置100、25、5、5、5、5 mL 的量瓶中,加甲醇稀釋并定容至刻度,制成系列濃度的混合對照品溶液液,精密吸取上述混合對照品儲備液和系列濃度的混合對照品溶液各10 μL,分別按“2.1”項下色譜條件進樣分析,以色譜峰面積響應值為縱坐標(y),相應的對照品質量濃度為橫坐標(x)進行線性回歸,得到的回歸方程及線性范圍見表2。

表2 4 種成分的線性回歸方程及線性范圍Tab 2 Linear regression equation and linearity of 4 components
2.6.2 精密度試驗 取“2.2”項下對照品溶液,按“2.1”項下色譜方法連續進樣6 次,計算朝藿定A、B、C 及淫羊藿苷的峰面積RSD值分別為1.1%、0.33%、0.41%和0.39%,表明儀器精密度良好。
2.6.3 穩定性試驗 精密吸取同一供試品溶液,按“2.1”項下色譜條件,分別在0、3、6、14、19、24 h 測定,結果朝藿定A、B、C 及淫羊藿苷的峰面積RSD值分別為0.51%、0.30%、0.37%和0.33%,表明供試品溶液在24 h 內穩定性良好。
2.6.4 重復性試驗 取淫羊藿配方顆粒供試品(批號:YYH02)溶液6 份,按“2.3”項下方法制備6 份供試品溶液,按“2.1”項下色譜條件進樣分析,以干燥品計,朝藿定A、B、C 及淫羊藿苷的含量分別為6.76、14.00、18.73、37.95 mg·g-1,RSD值分別為1.5%、0.81%、0.93%和0.69%,表明該方法重復性良好。
2.6.5 加樣回收試驗 取淫羊藿配方顆粒(批號:YYH02)適量,研細,取約0.125 g,精密稱定,按樣品與對照品(朝藿定A、B、C 及淫羊藿苷)的含量比為1∶0.5、1∶1 及1∶1.5 的比例加入朝藿定A、B、C 及淫羊藿苷對照品,每種比例平行3 份,按“2.3”項下方法制備9 份供試品溶液,按“2.1”項下色譜條件進樣分析,結果朝藿定A、B、C 及淫羊藿苷的加樣回收率分別在95.84%~99.78%、95.49%~101.62%、95.17%~101.41%、95.07%~99.92%,RSD值均小于2.0%,表明該分析方法準確度良好。
2.6.6 相對校正因子計算 根據“2.6.1”項下線性考察結果,以對照品的濃度及其對應的色譜峰面積,計算內標物淫羊藿苷與朝藿定A、B、C的相對校正因子[11]。朝藿定A、B、C 的平均相對校正因子分別為1.35、1.26 和1.24,RSD值分別為0.43%、1.1%和1.2%。
2.6.7 耐用性考察 精密吸取“2.2”項下對照品溶液,按“2.1”項下色譜條件測定,分別考察Waters ARC、ThermoU3000、Agilent 1260 infinity 3 種高效液相色譜儀和Agilent ZORBAX SB C18(4.6 mm×250 mm,5 μm)、Phenomenex Luna C18(4.6 mm×250 mm,5 μm)、Waters XSelect HSS T3(4.6 mm×250 mm,5 μm)3 種不同色譜柱,以及不同柱溫和流速對朝藿定A、B、C 的相對校正因子以及各色譜峰分離度的影響。結果表明,不同色譜儀、色譜柱、柱溫及流速下測定的朝藿定A、B、C 的相對校正因子RSD值均小于2.0%,且各色譜峰的分離度均大于1.5,表明該方法耐用性較好。
2.6.8 色譜峰定位 根據“2.6.7”項下耐用性考察結果,以淫羊藿苷峰為參照峰,分別計算Waters ARC、Thermo U3000、Agilent 1260 infinity 3 種高效液相色譜儀和Agilent ZORBAX SB C18(4.6 mm×250 mm,5 μm)、Phenomenex Luna C18(4.6 mm×250 mm,5 μm)、Waters XSelect HSS T3(4.6 mm×250 mm,5 μm)3 種不同色譜柱,以及不同柱溫和流速對朝藿定A、B、C 相對于參照峰的相對保留時間值。結果顯示,不同的儀器、色譜柱、柱溫和流速對朝藿定A、B、C 的相對保留時間影響較小,RSD均小于5.0%,因此,可以采用相對保留時間作為色譜峰定位的依據,朝藿定A、B、C 的相對保留時間值取不同儀器、色譜柱、柱溫和流速下相對保留時間的均值,分別是0.71、0.79 和0.88。
2.6.9 QAMS 與EMS 比較 取12 批淫羊藿配方顆粒樣品,按“2.3”項下方法制備供試品溶液,按“2.1”項下色譜條件進樣分析,分別采用外標法(ESM)和QAMS 計算12 批淫羊藿配方顆粒中朝藿定A、B、C 的含量,并計算兩種測定法的相對平均偏差(RAD)(見表3),結果顯示,兩種方法測定的朝藿定A、B、C 的RAD 均小于1.0%,說明兩種方法計算結果一致,一測多評法能準確測定4 種成分的含量。12 批淫羊藿配方顆粒淫羊藿苷的含量均大于20 mg·g-1,12 批淫羊藿配方顆粒總黃酮醇苷的含量在46.73~88.94 mg·g-1。
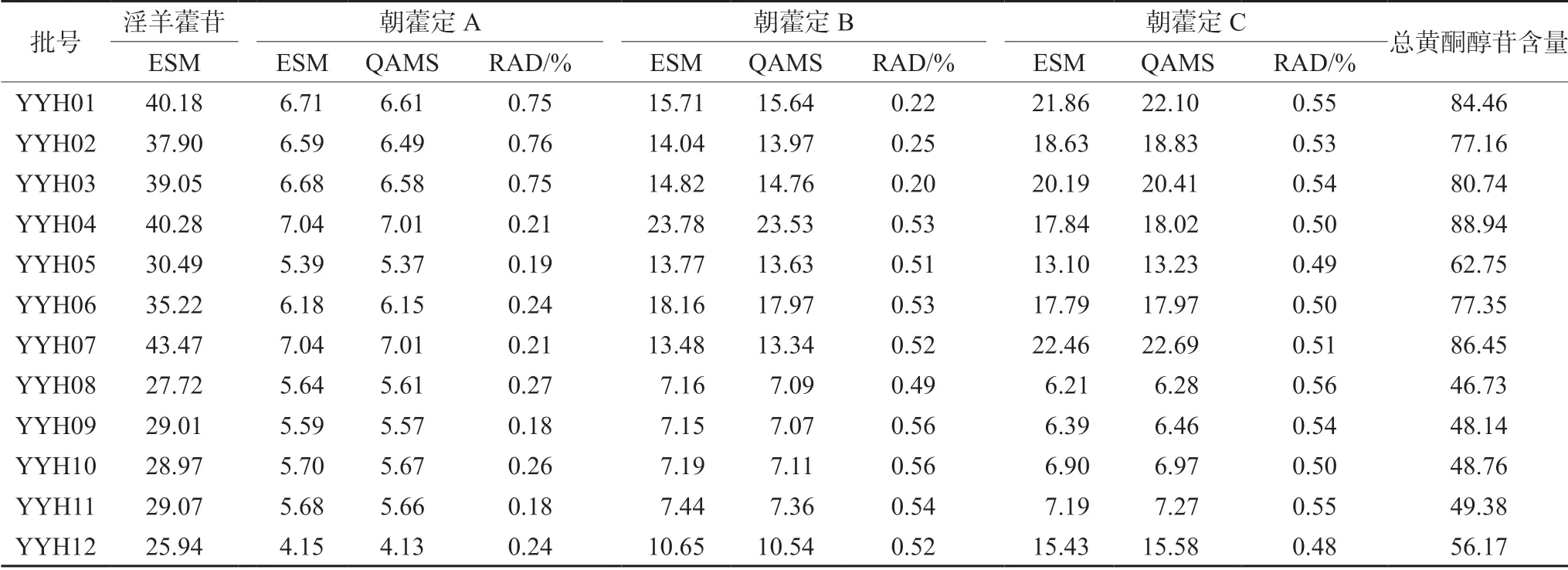
表3 12 批淫羊藿配方顆粒含量測定結果(以干燥品計,mg·g-1)Tab 3 Determination of 12 batches of Herba epimedii dispensing granules (mg·g-1)
3 討論
3.1 供試品溶液的制備
以“特征圖譜總峰面積/稱樣量”及總黃酮醇苷的含量為指標,系統考察了提取溶劑(不同比例的甲醇-水、乙醇-水、甲醇和乙醇)、提取方式(超聲和回流兩種不同方式)和提取時間(15、30、45、60 min)對淫羊藿配方顆粒特征圖譜及含量測定的影響,結果顯示,以75%乙醇50 mL超聲處理(功率250 W,頻率40 kHz)45 min 效果最佳。
3.2 特征圖譜和一測多評研究
對12 批淫羊藿配方顆粒特征圖譜研究,確定了7 個共有特征峰,指認了其中5 個,各特征峰的相對保留時間一致性較好,相對峰面積差異較大,但指紋圖譜的整體,相似度較高,都大于0.9。含量測定結果顯示,4 種成分的含量在不同批次樣品間相差較大,可能與原料的來源和生產工藝的不同有關;外標法與一測多評法所得結果無明顯差異,表明本研究建立的一測多評法能夠代替外標法對淫羊藿配方顆粒進行質量控制。
4 小結
中藥化學成分繁多,單一成分難以準確評價配方顆粒的質量,一測多評法是多指標質量控制方法之一,將一測多評法與指紋/特征圖譜法相結合,從定性、定量兩個方面來評價淫羊藿配方顆粒的質量,該方法快速準確,簡便高效,專屬性良好,為淫羊藿配方顆粒的質量控制提供參考。
猜你喜歡
數學小靈通·3-4年級(2024年2期)2024-05-15 02:02:28
中學生數理化(高中版.高考數學)(2022年3期)2022-04-26 14:04:16
數學年刊A輯(中文版)(2020年1期)2020-05-19 00:30:36
空間科學學報(2020年2期)2020-04-01 03:50:40
瘋狂英語·新策略(2019年10期)2019-12-13 08:43:28
中等數學(2019年8期)2019-11-25 01:38:14
當代陜西(2019年10期)2019-06-03 10:12:04
新聞傳播(2018年11期)2018-08-29 08:15:24
數學小靈通·3-4年級(2017年9期)2017-10-13 08:10:54
廣西科技大學學報(2016年1期)2016-06-22 13:10:38
