肉蓯蓉總苷中4種雌激素樣活性成分的藥動學研究
2021-08-23 03:30:56揚高佳雪綦崢徐蓓蕾曲中原梁偉宋輝孫向明丁振鐸李文蘭
中成藥 2021年8期
關鍵詞:血漿
胡 揚高佳雪綦 崢徐蓓蕾曲中原梁 偉宋 輝孫向明丁振鐸 李文蘭
(哈爾濱商業大學,黑龍江 哈爾濱150076)
肉蓯蓉作為補益中藥,可補腎虛、壯腎陽、益精血,在我國用藥歷史悠久,臨床常用于治療女子宮寒不孕、男子腎虛陽痿等癥,同時具有保肝護肝、抗氧化、抗衰老、抗腫瘤作用,有“沙漠人參”的美譽[1?2]。中藥藥代動力學是以遵循中醫藥理論為前提,對藥物進入機體后的時量?時效關系及其動態變化規律,通過數學函數進行定量描述的一門學科[3],隨著中藥現代化的不斷推進、中醫藥理論的蓬勃發展,當前中藥藥代動力學的研究對象已逐漸由傳統的單體成分轉變為具有明顯藥效作用的活性成分群[4?5],由于中藥具有多成分、多靶點、整合調節的特點[6?7],這種轉變可使得相關研究更透徹,更具有科學意義。
近年來,課題組對肉蓯蓉雌激素作用進行了大量研究,證實其有效部位為總苷[8],并通過構建“固有成分?體內成分?雌激素活性”譜效研究模型,確定松果菊苷、丁香苷、紅景天苷、毛蕊花糖苷等13種成分是相關藥效物質基礎[9]。為進一步揭示上述成分的體內過程,本實驗采用超高效液相色譜?串聯三重四級桿質譜(UPLC?MS/MS)對松果菊苷、丁香苷、紅景天苷、毛蕊花糖苷進行藥動學研究,以期將肉蓯蓉深度開發為植物雌激素5 類新藥奠定基礎。
1 材料
1.1 試劑與藥物 肉蓯蓉總苷(質量分數為62.5%,以毛蕊花糖苷計),按照文獻[10]報道的方法自制。綠原酸、紅景天苷、毛蕊花糖苷、松果菊苷、丁香苷對照品(純度>98%,批號分別為110753?201716、110818?201206、111530?201411、111670?201304、11574?200201)均購自中國食品藥品檢定研究院。乙腈、甲醇為色譜純(德國默克公司,批號分別為20170410、20170311);甲酸為色譜純(河南科銳化工有限公司,批號20160509);正丁醇為分析純(批號20140210)、乙酸乙酯(批號20150308)為分析純(天津福晨化學試劑有限公司);水為純凈水[屈臣氏集團(香港)有限公司,批號20171205]。
1.2 儀器 ACQUITY UPLC 色譜儀、Xevo TQD 質譜儀(美國Waters 公司);AR1140 電子分析天平(美國奧豪斯公司);超聲波清洗器、Neofuge 高速冷凍離心機(上海力申科學儀器有限公司);GENIUS N118LA 氮氣發生器(英國Peak 公司)。
1.3 動物 Wistar 雌性大鼠,體質量(210±20)g,購自哈爾濱醫科大學實驗動物學部,動物生產許可證號SCXK(黑)2013?001,自由飲水飲食,在室溫(23±2)℃、相對濕度50%~60%下適應性飼養7 d。
2 方法與結果
2.1 溶液制備
2.1.1 對照品溶液 精密稱取松果菊苷、丁香苷、紅景天苷、毛蕊花糖苷對照品適量,50% 乙腈稀釋,即得(質量濃度為250 μg/mL)。
2.1.2 內標溶液 精密稱取綠原酸對照品適量,50%乙腈稀釋,即得(質量濃度為500 ng/mL)。
2.1.3 灌胃液 取肉蓯蓉總苷適量,蒸餾水稀釋至75 mg/mL(含松果菊苷1.346 mg/mL、丁香苷1.195 mg/mL、紅景天苷0.631 mg/mL、毛蕊花糖苷1.487 mg/mL),即得。
2.2 分析條件
2.2.1 色譜 Waters BEH C18色譜柱(1.7 μm,
2.1 mm×100 mm);流動相乙腈?水(含0.1%甲酸),梯度洗脫,程序見表1;體積流量0.2 mL/min;柱溫25.4 ℃;進樣量5 μL。

表1 梯度洗脫程序Tab.1 Gradient elution programs
2.2.2 質譜 電噴霧離子源(ESI),多反應監測模式(MRM);錐孔電 壓50 V;毛細管電壓3.0 kV;錐孔氣體積流量50 L/h;霧化氣體積流量650 L/h,溫度350 ℃;源溫度150 ℃。定量離子對、參數見表2。

表2 各成分定量離子對、參數Tab.2 Quantitative ion pairs and parameters for various constituents
2.3 分組、給藥及樣品采集 10 只大鼠隨機分為給藥組、空白組,每組5 只,灌胃給藥,劑量為1.35 g/(kg·d-1)(以肉蓯蓉總苷計),空白組給予等體積蒸餾水,共6 次,每次間隔12 h,取血前12 h 禁食,自由飲水。于末次給藥后0.25、0.5、1、1.5、2、4、6、8、10、12 h 眼眶靜脈叢取血約500 μL 至肝素化EP 管中,靜置0.5 h 后12 000 r/min 離心10 min,吸取上層血漿,-20 ℃下保存。同法制備空白血漿。
2.4 血漿樣品預處理 將“2.1.1”項下對照品溶液稀釋至500 ng/mL,取3 份,每份20 μL,置于EP 管中,40 ℃氮氣吹干,加入200 μL 空白血漿、20 μL 內標溶液,渦旋30 s 混合均勻,加入乙酸乙酯、甲醇、水飽和正丁醇各660 μL,渦旋混合3 min,12 000 r/min 離心10 min,取上清液,40 ℃氮氣吹干,200 μL 60%甲醇復溶,渦旋1 min,經0.22 μm 微孔濾膜過濾,在“2.2”項條件下進樣測定。待測成分與綠原酸峰面積之比記為A;取200 μL 空白血漿3 份,加入20 μL 內標溶液,渦旋30 s 混勻,加入乙酸乙酯、甲醇、水飽和正丁醇各660 μL,渦旋混合3 min,12 000 r/min 離心10 min,取上清液,40 ℃氮氣吹干,加入500 ng/mL對照品溶液20 μL,渦旋1 min,40 ℃氮氣吹干,200 μL 60%甲醇復溶,渦旋1 min,0.22 μm 微孔濾膜過濾,在“2.2”項條件下進樣測定。待測成分與綠原酸峰面積之比記為B,以A/B計算提取回收率,結果見表3,可知方法2 中各成分提取回收率均較高,故選擇甲醇沉淀蛋白法進行血漿樣品預處理。
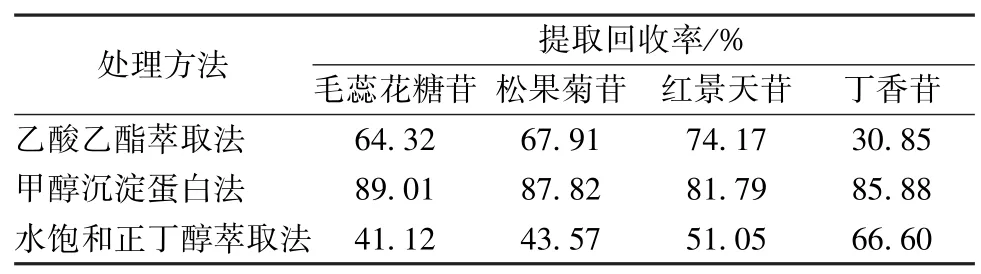
表3 不同預處理方法下提取回收率(n=3)Tab.3 Extract recoveries under different pretreatment methods(n=3)
2.5 方法學考察
2.5.1 專屬性試驗 按“2.4”項下最優方法制備空白血漿供試品溶液(不加對照品、內標溶液)、模擬血漿供試品溶液(空白血漿中加入500 ng/mL 對照品、內標溶液)、給藥60 min 后血漿供試品溶液(加內標溶液),在“2.2”項條件下進樣測定,質譜圖見圖1~2,可知正離子模式下丁香苷m/z395.10 峰的保留時間為2.300 min,綠原酸m/z355.10 峰的保留時間為2.471 min;負離子模式下紅景天苷m/z299.10 峰的保留時間為2.191 min,綠原酸的m/z353.00 峰的保留時間為2.476 min,毛蕊花糖苷m/z623.20 峰的保留時間為3.210 min,松果菊苷m/z785.30 峰的保留時間為2.640 min。多反應監測(MRM)離子色譜圖見圖3~4,可知血漿中內源性物質、代謝產物對成分測定無干擾,表明該方法專屬性良好。
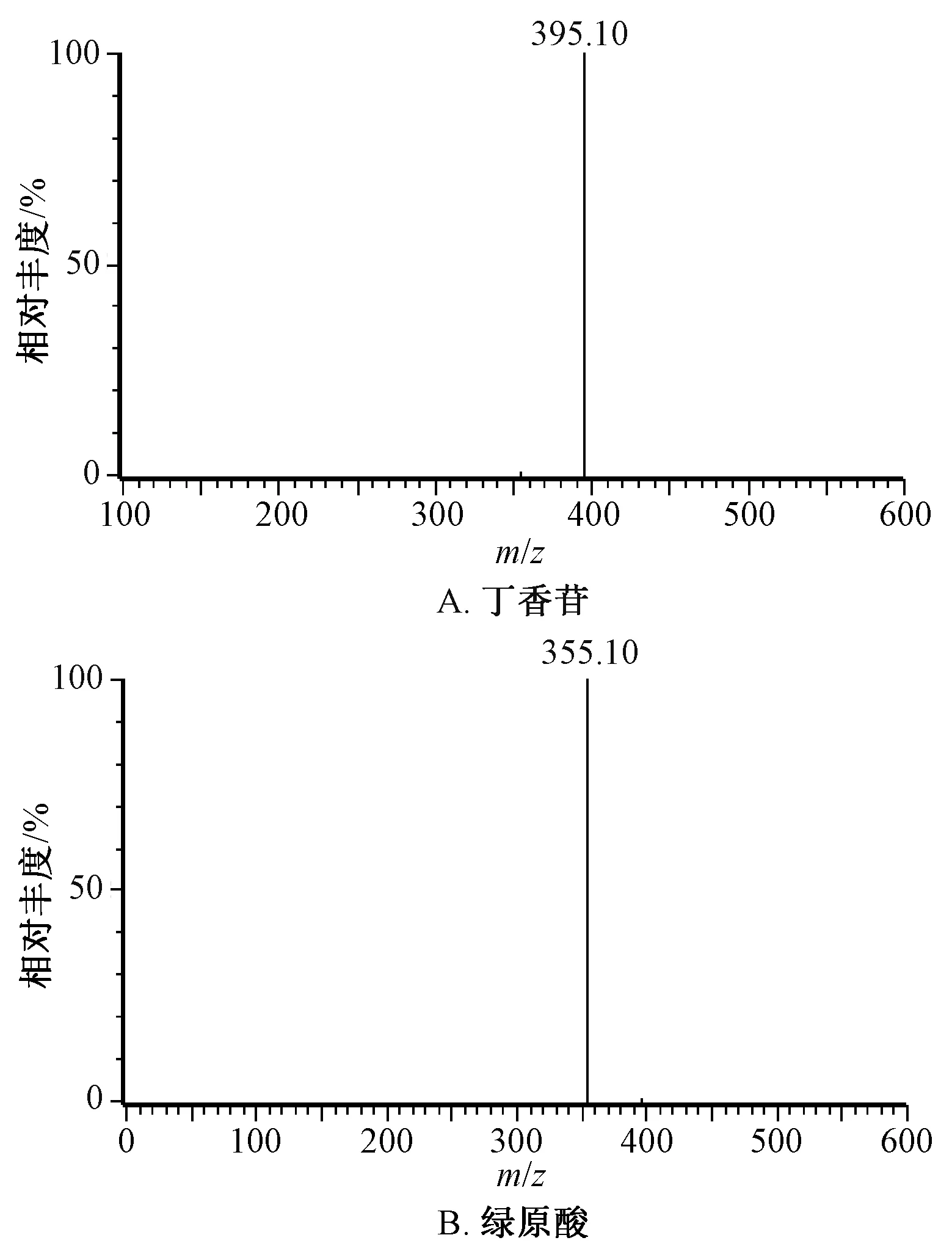
圖1 各成分質譜圖(正離子模式)Fig.1 Mass spectrograms of various constituents(positive ion mode)
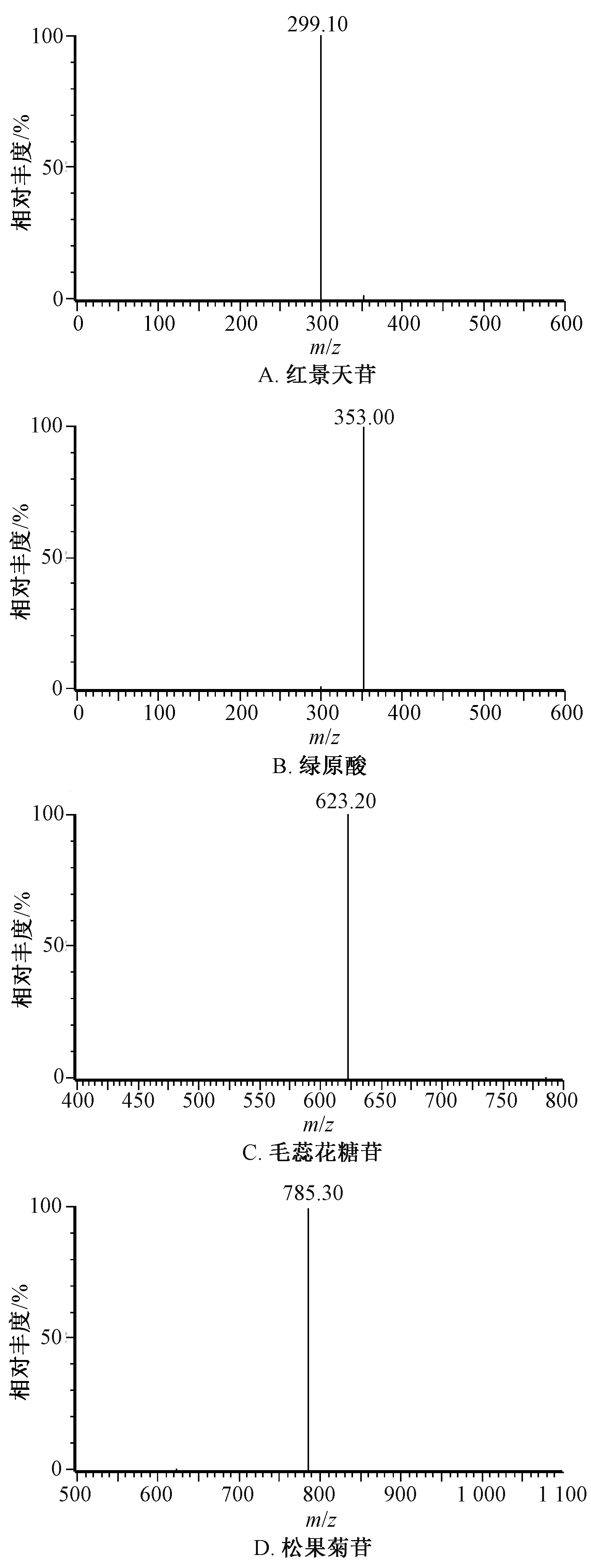
圖2 各成分質譜圖(負離子模式)Fig.2 Mass spectrograms of various constituents(negative ion mode)
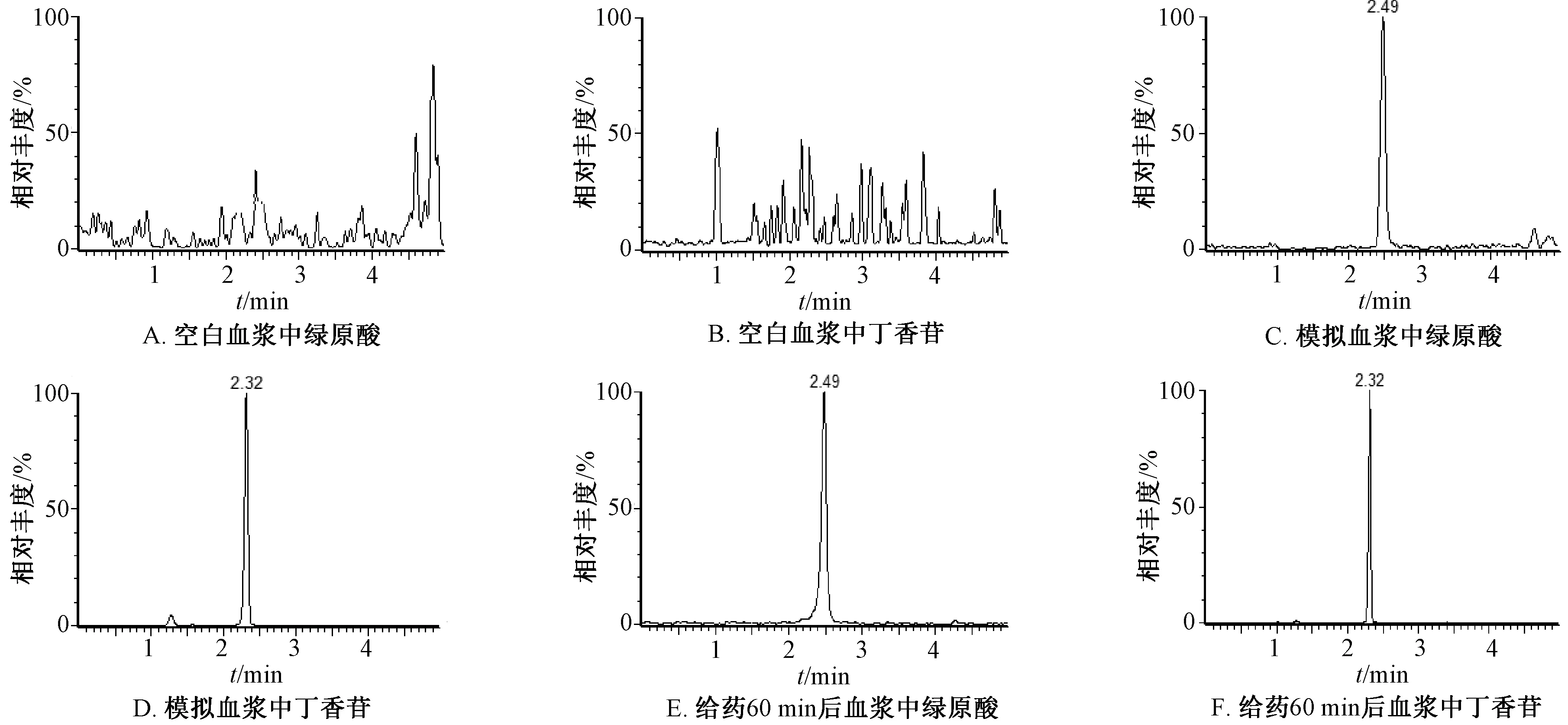
圖3 各成分MRM 色譜圖(正離子模式)Fig.3 MRM chromatograms of various constituents(positive ion mode)
2.5.2 線性關系考察 將“2.1.1”項下對照品溶液稀釋至1、10、100、200、500、1 000、2 500 ng/mL,各取20 μL,40 ℃氮氣吹干,加入200 μL 空白血漿、20 μL 內標溶液,按“2.4”項下方法處理,分別制備成質量濃度為0.1、1、10、20、50、100、250 ng/mL的溶液,在“2.2”項條件下進樣測定,平行3 次。以各成分質量濃度為橫坐標(X),待測成分、綠原酸峰面積比值為縱坐標(Y)進行回歸,以S/N =10 時的樣品質量濃度為定量限,結果見表4,可知在各自范圍內線性關系良好。
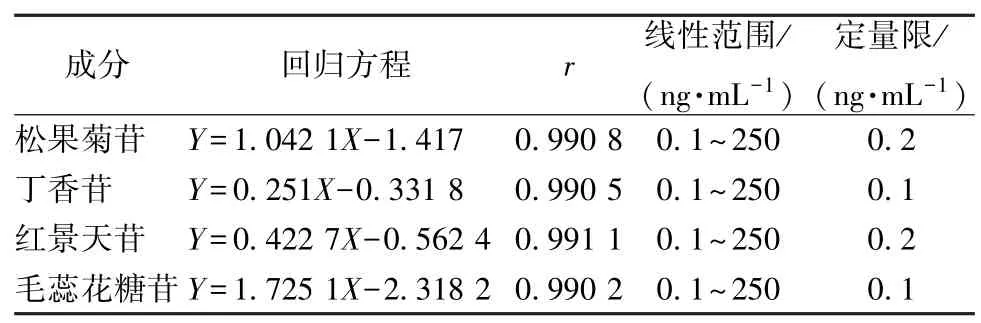
表4 各成分線性關系Tab.4 Linear relationships of various constituents
2.5.3 精密度、準確度試驗 按“2.5.2”項下方法制備0.5、20、200 ng/mL 模擬血漿供試品溶液作為質控樣品,平行6 份,每天繪制1 次標準曲線,完成1個批次測定,以當日標準曲線計算質控樣品質量濃度,連續6 d,結果見表5。由此可知,各成分日內、日間精密度RSD 為1.62%~6.38%,RE 為-6%~19.7%,滿足生物樣品定量分析要求。
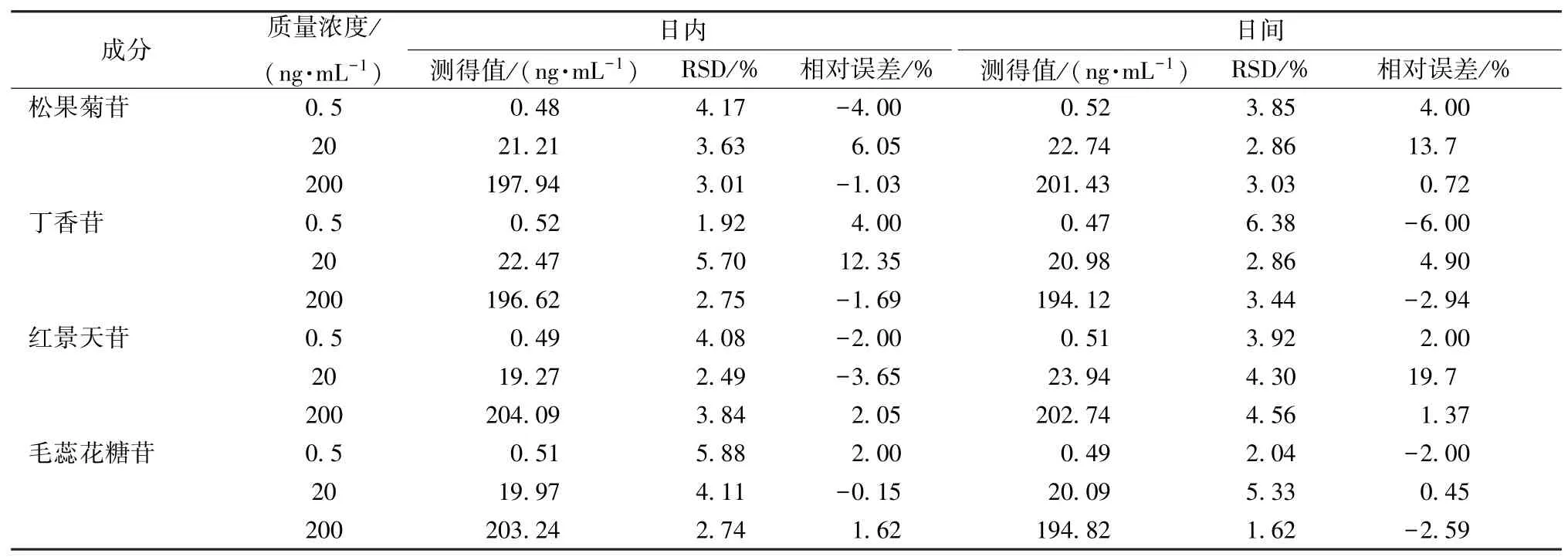
表5 各成分精密度、準確度試驗結果(n=6)Tab.5 Results of precision and accuracy tests for various constituents(n=6)
2.5.4 基質效應、提取回收率試驗 取20 μL 內標溶液,40 ℃氮氣吹干,加入200、20、0.5 ng/mL對照品溶液各200 μL,濾過,在“2.2”項條件下進樣測定,待測成分、綠原酸峰面積之比計為Al;取200 μL 空白血漿,加入660 μL 甲醇,渦旋混合3 min 后12 000 r/min 離心10 min,取上清液,40 ℃氮氣吹干,加入20 μL 內標溶液,40 ℃氮氣吹干,加入3個質量濃度對照品溶液各200 μL,渦旋1 min,濾過,在“2.2”項條件下進樣測定,待測成分、綠原酸峰面積之比計為A2;取3個質量濃度對照品溶液各200 μL,加入20 μL 內標溶液,40 ℃氮氣吹干,再加入空白血漿200 μL、甲醇660 μL,渦旋混合3 min,12 000 r/min 離 心10 min,取上清液,40 ℃氮氣吹干,200 μL 60%甲醇復溶,渦旋1 min,濾過,在“2.2”項條件下進樣測定,待測成分、綠原酸峰面積之比計為A3,平行6 次,以A2/A1計算基質效應,A3/A2計算提取回收率,結果見表6。由此可知,該方法無明顯基質效應,提取回收率較高,符合生物樣品分析要求。
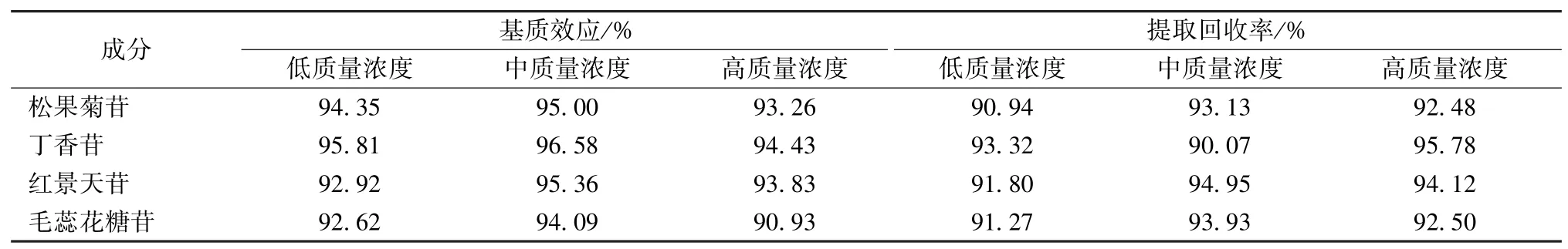
表6 各成分基質效應、提取回收率試驗結果(n=6)Tab.6 Results of matrix effect and extraction recovery tests for various constituents(n=6)
2.5.5 穩定性試驗
2.5.5.1 室溫穩定性 取“2.5.3”項下質控樣品,室溫下放置6 h,在“2.2”項條件下進樣測定。
2.5.5.2 冷凍穩定性 取“2.5.3”項下質控樣品,-20 ℃下凍存7 d,在“2.2”項條件下進樣測定。

圖4 各成分MRM 色譜圖(負離子模式)Fig.4 MRM chromatograms of various constituents(negative ion mode)
2.5.5.3 凍融穩定性 取“2.5.3”項下質控樣品,-20 ℃下凍存24 h 后取出,室溫下完全自然解凍,在“2.2”項條件下進樣測定,再放回-20 ℃下凍存24 h 以上,重復3 次。
2.5.5.4 結果分析 上述3種試驗平行6 次,結果見表7。由此可知,不同條件下各成分含量RSD均小于10%,表明樣品穩定性良好,但仍應盡量減少凍融次數。

表7 各成分穩定性試驗結果(n=6)Tab.7 Results of stability tests for various constituents(n=6)
2.6 藥動學研究 將“2.3”項下血漿樣品按“2.4”項下最優方法處理后,在“2.2”項條件下進樣測定,通過PK Solver 2.0 軟件的非房室模型繪制血藥濃度?時間曲線,計算主要藥動學參數,結果見圖5、表8。
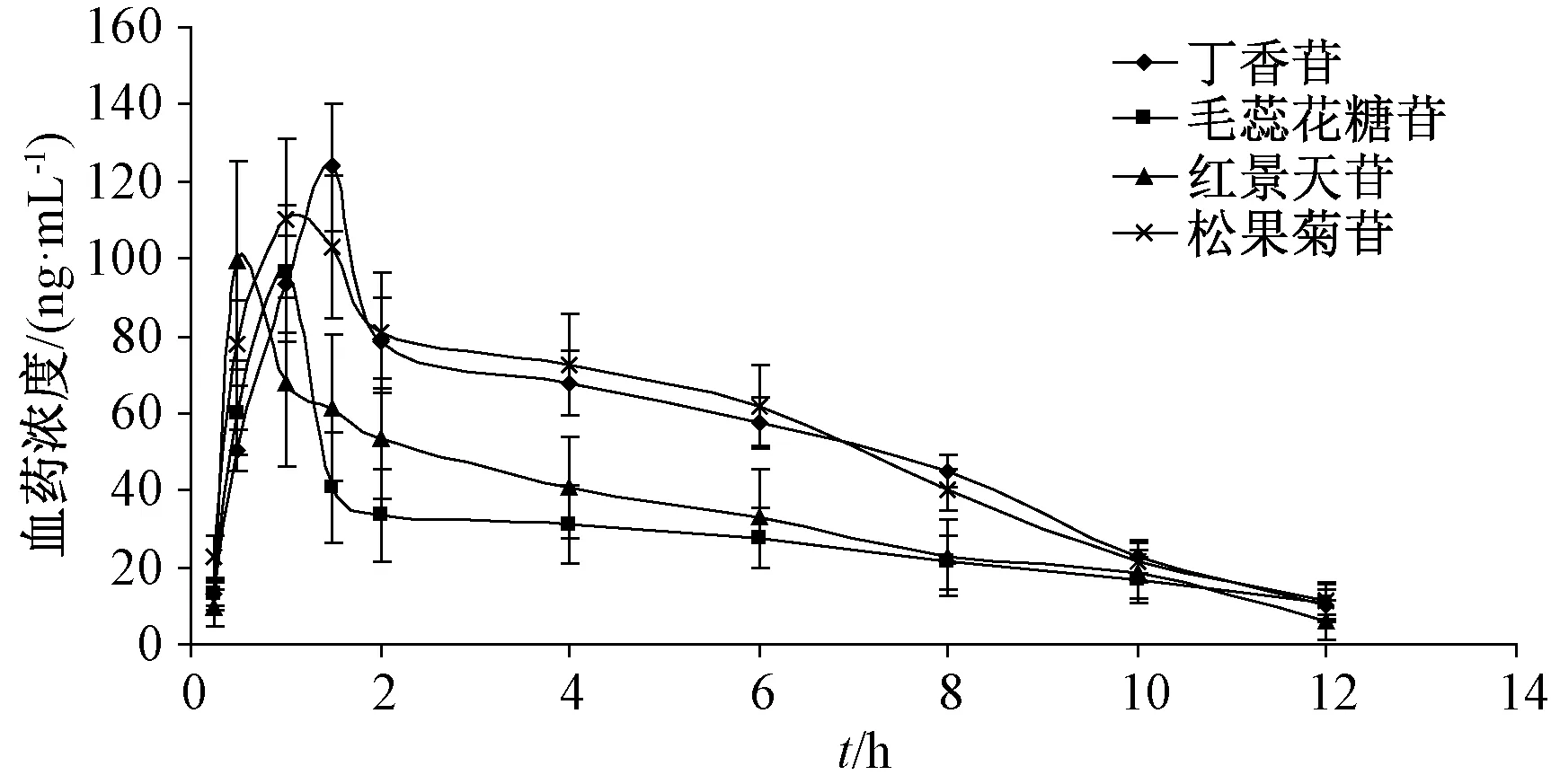
圖5 各成分血藥濃度?時間曲線Fig.5 Plasma concentration?time curves for various constituents
表8 各成分主要藥動學參數(, n=5)Tab.8 Main pharmacokinetic parameters for various constituents(, n=5)

表8 各成分主要藥動學參數(, n=5)Tab.8 Main pharmacokinetic parameters for various constituents(, n=5)
3 討論與結論
肉蓯蓉雌激素作用是現代藥理研究成果[11?13],課題組前期發現,肉蓯蓉總苷中松果菊苷、丁香苷、紅景天苷、毛蕊花糖苷等13種成分是關鍵物質[9],其中毛蕊花糖苷、松果菊苷是2020年版《中國藥典》 肉蓯蓉含量測定項下指標成分[14]。以此為基礎,本實驗選擇松果菊苷、丁香苷、紅景天苷、毛蕊花糖苷進行大鼠體內藥動學研究,以期揭示肉蓯蓉雌激素作用活性物質在體內的動態變化過程,整體實驗思路與中藥作用特點相契合,對傳統中醫藥理論賦予了更現代、更科學的詮釋,不僅有助于中藥質量控制和評價方法的完善,而且對臨床合理用藥、新藥研發具有重要意義。
本實驗采用UPLC?MS/MS 法檢測松果菊苷、丁香苷、紅景天苷、毛蕊花糖苷血藥濃度,其超低檢測限更符合體內樣品含量較低的實際檢測需求。儀器靈敏度往往也是影響藥動學實驗結果的重要因素之一[15],與傳統的HPLC 法相比,UPLC?MS/MS 法優勢不僅體現在靈敏、快速、高效、節約成本,最重要的是數據更準確,更能真實反映雌激素活性成分血藥濃度的動態變化過程。結果顯示,給藥后各成分在30~90 min 之間均達到最大血藥濃度,其中丁香苷、松果菊苷下降迅速,半衰期在2 h左右,而毛蕊花糖苷、紅景天苷相對較慢,半衰期在4~5 h,整體表現為吸收、消除速度均較快,以期為肉蓯蓉雌激素作用的深入研究提供依據。
猜你喜歡
現代臨床醫學(2022年4期)2022-09-29 07:38:00
昆明醫科大學學報(2021年4期)2021-07-23 01:21:50
天津醫科大學學報(2019年6期)2019-08-13 07:04:34
云南醫藥(2019年3期)2019-07-25 07:25:14
現代檢驗醫學雜志(2016年5期)2016-08-20 03:16:56
海南醫學(2016年8期)2016-06-08 05:43:00
西南軍醫(2016年5期)2016-01-23 02:20:33
川北醫學院學報(2015年5期)2015-12-05 08:22:28
醫學研究雜志(2015年9期)2015-07-01 17:28:15
現代檢驗醫學雜志(2015年1期)2015-02-06 01:59:26
