氯代硝基苯選擇性加氫催化劑研究現狀與進展
2021-08-21 07:32:46王敏嘉周少東阮建成李嶸嶸陳新志
化工進展 2021年8期
關鍵詞:催化劑
王敏嘉,周少東,阮建成,李嶸嶸,陳新志
(1浙江大學化學工程與生物工程學院;浙江省化工高效制造技術重點實驗室,浙江杭州 310027;2浙江大學衢州研究院,浙江衢州 324000;3臺州學院浙江省醫藥化工廢物回收利用綜合工程研究中心,浙江臺州 318000)
鹵代芳胺是工業上合成許多精細化學品的重要中間體,廣泛應用于染料、藥物、除草劑和殺蟲劑。合成方法主要有化學還原法、電化學還原法和催化加氫還原法,催化加氫法因綠色、高效等優勢成為近年來研究的熱點,而如何實現高效的選擇性催化又是催化劑設計的難點[1]。
硝基芳烴的加氫反應途徑如圖1所示,其中,亞硝基化合物和羥胺是中間體,而一系列副反應可能導致亞胺、偶氮苯等副產物的形成。在硝基芳烴加氫中,選擇性很難控制,反應物中存在一個及以上的可還原基團時,加氫通常優先發生在非目標基團上,得到低收率的所需官能化苯胺[2]。以CAN為例,由于具有給電子特性(相對于吸電子的—Cl和—NO2基團)的—NH2基團可通過苯環誘導電子轉移,增加—C1的極化,促進解離氫攻擊C—Cl鍵,導致“脫鹵現象”。脫鹵副反應不僅降低了反應選擇性,還使催化劑中毒、降低其活性和壽命,其產生的鹵化氫不僅使金屬活性組分流失,也會腐蝕設備、造成安全隱患,這對選擇性催化劑的設計提出了更高的要求。
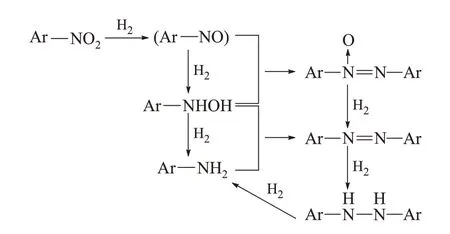
圖1 硝基加氫反應過程
1 多相催化劑
多相催化劑具有活性高、易分離的特點,工業上廣泛應用。多相金屬催化劑性能的改變通常與金屬相分散度、活性中心的調變和載體性質等有關。因此,還原多官能有機化合物的選擇性取決于反應基團對表面結構的不同敏感性。
1.1 調變金屬相分散度
負載金屬的粒度是決定催化劑性能的關鍵因素,除可能導致的物理化學性質的變化外,其表面效應和尺寸效應也是影響催化性能的重要原因。
Chen等[3]通過溶膠-凝膠法將還原性有機基團引入到二氧化硅中后,創造了一個新的原位還原法,實現了一步完成HAuCl4的引入和還原,得到了Au/SiO2催化劑,金納米顆粒均勻分布在7.0~9.0nm的范圍內,在適當反應條件下,CNB轉化率、CAN選擇性均接近100%。
貴金屬團簇催化劑因其高原子利用率和優異的催化活性而被廣泛研究,但其易聚集性限制了它們的應用。通過構建共價有機框架物(covalent organic frameworks,COFs)可有效控制貴金屬團簇的尺寸,提高其催化性能。Fan等[4]利用三嗪基官能化的共價有機框架(三嗪基-COFs)的限制效應來錨定鈀團簇和控制其尺寸。他們通過三聚氯氰分別與對苯二胺和4,4'-二氨基聯苯縮合合成共價有機框架COF-Ph和COF-BPh,然后通過雙溶劑法制備了Pd@COF-Ph和Pd@COF-BPh催化劑(圖2),鈀團簇固定在COFs孔中,尺寸為0.8nm。所得催化劑對硝基芳烴的還原表現出極高的催化活性和穩定性,其中,CAN的收率可達99%,且不發生脫氯。其作者認為,COFs孔的限制作用提高了原子利用率,即增強了催化劑的活性,而選擇性的提高則歸因于三嗪基上氮原子對鈀原子的電子效應。
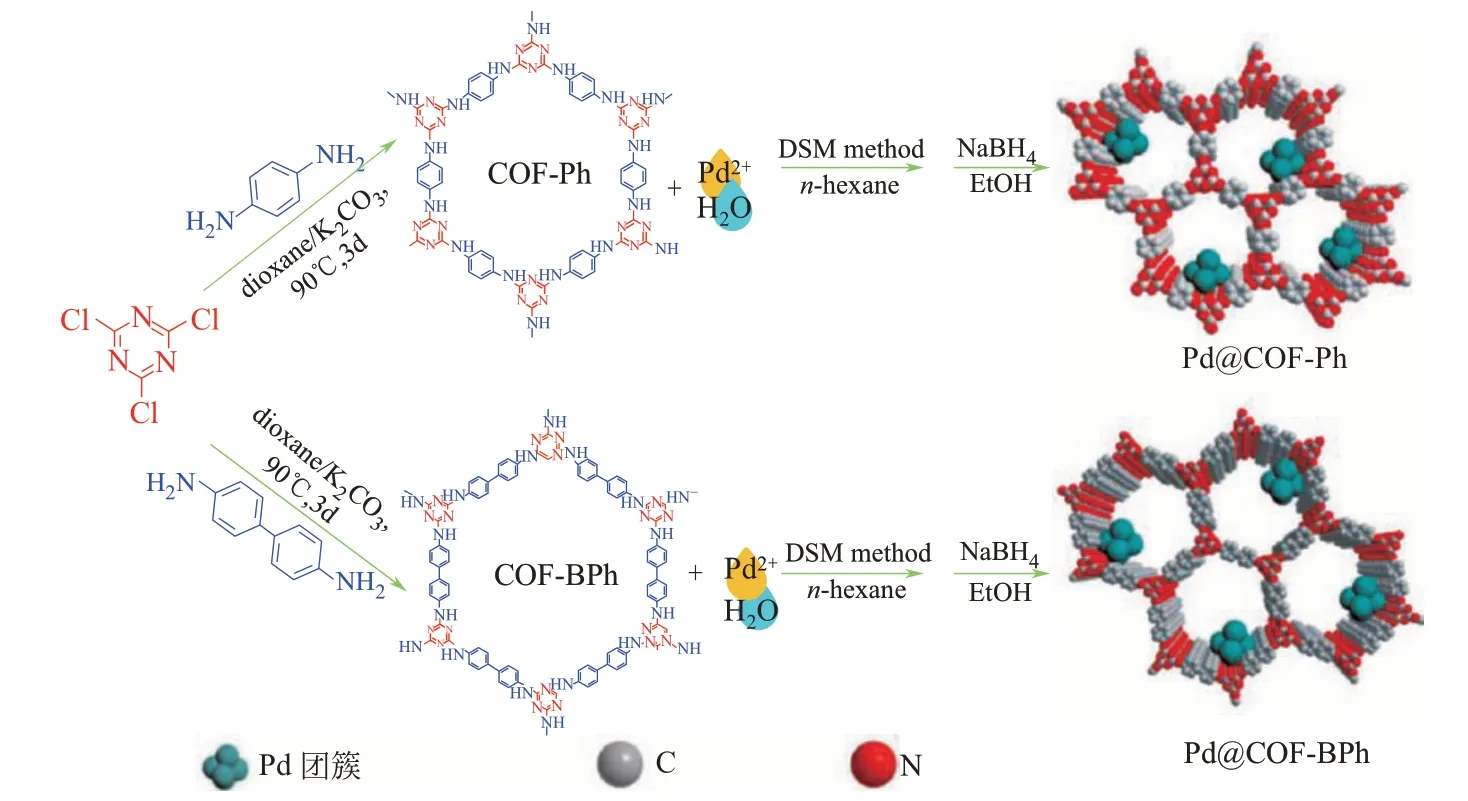
圖2 Pd@COF-Ph和Pd@COF-BPh催化劑的制備[4]
原子級分散的金屬尺寸限制低,可最大限度地利用原子,同時減少貴金屬的消耗,單原子催化劑(SACs)因其獨特的催化性能而備受關注。Wei等[5]以H2PtCl6和Fe(NO3)3為金屬前體,Na2CO3為沉淀劑,在50℃共沉淀法制備了一系列Pt/FeOx催化劑。通過改變鉑負載量和還原溫度,鉑金屬的形態發生從孤立的單原子、松散結合的團簇、亞納米簇到三維納米粒子的變化。單原子Pt/FeOx催化劑在較低還原溫度(200℃)下得到。作者指出,相較于其他結構,單原子結構帶更多的正電荷,有利于硝基的優先吸附,且單原子是化學選擇性還原最活躍的位點。將其應用在o-CNB加氫上,轉化率可達100%,主產物選擇性可達97.4%,重復使用至少5次而沒有任何選擇性損失。
1.2 載體的選擇與改性
載體的性質通常能影響催化劑金屬相的分散度或活性。Xiong等[6]考察了載體(SiO2、ZrO2、TiO2、Al2O3)對鎳催化劑性能的影響。其中,Ni/TiO2的催化性能最好,o-CNB轉化率和選擇性均可達99%以上。作者認為在高溫還原條件下,低價氧化物TiOx(x<2)和Ni的強金屬載體相互作用(strong metal-support interactions,SMSI)使系統表面自由能降低,從而發生TiOx到Ni表面的部分遷移,產生的氧空位可以與N==O基團中的氧原子配位,使得N==O鍵被極化,故易被吸附在鎳顆粒上的氫攻擊。
Wang等[7]設計了以γ-磷化鋯(γ-ZrP)為載體的鉑催化劑,對CNB選擇性加氫顯示出優異的活性和選擇性,在原料完全轉化時沒有任何脫氯。作者認為ZrP的—OH基團抑制了CAN的—NH2基團的給電子能力,因此CAN的脫氯被抑制;而且ZrP載體和鉑顆粒之間的SMSI作用減弱了鉑原子向芳香環的電子反饋程度,進一步抑制了C—Cl鍵的氫解。
調變載體的表面性質可有效改善催化劑性能,如利用載體上的缺陷位/官能團來錨定活性位點。Wu等[8]制備了Pt/C催化劑,用硝酸處理活性炭后,負載的鉑納米粒子的表面變得能暴露更多的Ptδ+物種。硝酸處理顯著增加了活性炭上包括羧基和酸酐基團等含氧官能團的數量,作者認為碳載體上的酸性基團是金屬納米粒子被錨定的位置,這些基團通過氫鍵與極性硝基相互作用,促進其在鉑納米粒子上的吸附。作者還在碳載體上引入了表面磷雜原子,當催化劑在較高溫度下被還原時,由于鉑和磷物種之間的相互作用,在負載的金屬顆粒的表面上形成Pt-POx復合物,這種復合物有助于選擇性吸附帶有極性硝基的底物,從而提高對硝基的選擇性加氫。
1.3 雙金屬協同催化
除了調變載體,作為負載型催化劑主體之一的金屬相也是改性的重點對象。Han等[9]研究了過渡金屬(Cr、Mn、Fe、Co、Ni和Cu)復合催化劑PtM/TiO2對CNB加氫性能的影響。其中,PtFe/TiO2顯示出最佳選擇性(98.0%)。作者提出當鉑與具有給電子特征的鐵合金化時,鐵通過在鉑活性位點誘導更高的電子密度來激活N==O鍵,從而促進反應速率。但當鐵含量高于1.0%時,催化活性急劇下降,原因可能是鉑活性中心被鐵部分覆蓋。Lihama等[2]也探究了一系列負載在二氧化硅上的鉑基雙金屬催化劑的性能,其中PtZn/SiO2催化劑的效果最佳,p-CNB的轉化率可達100%,主產物p-CAN的選擇性可達99%,他們也歸納出類似的結論,即隨著鉑位點上電子密度的增加,由于CAN上氯的吸電子性以及氨基的強給電子性,CAN與富電子的鉑位點產生靜電排斥作用,從而使脫氯被抑制。
Cárdenas-Lizana等[10]通過共沉積-沉淀/共浸漬制備了Au-Pd/Al2O3催化劑,在金體系中使用鈀作為促進劑導致雙金屬Au-Pd顆粒的形成。表征結果表明存在雙金屬顆粒和表面金-鈀相互作用,促成了高反應速率和高選擇性。
Cheng等[11]通過簡單還原CoNiAl-LDH前體制備了包埋在Al2O3納米片中的CoNi合金納米粒子,并將其用于以氨硼烷為氫源的硝基芳烴加氫反應,其中Co0.67Ni0.33/Al2O3表現出最佳催化性能,p-CAN的收率可達99%。這可歸因于CoNi合金的結構優化了純組分表面的電子結構,合金體系的d帶中心勢能比Ni(111)和Co(111)的都低,這就導致了它們吸附和活化能壘的差異,故CoNi合金不僅有利于促進氨硼烷的水解,且優化了中間體的吸附性能,進一步促進了硝基芳烴對胺的選擇性還原。
Wang等[12]通過封裝法制備了一種在雙殼碳載體中捕獲Pd-Fe雙金屬納米粒子的管狀催化劑(見圖3),合成的CNT/PdFe/NC催化劑對硝基芳烴還原反應比相應的單金屬催化劑有更好的催化活性,從而顯示出雙金屬組分增強的協同效應。根據表征結果,作者認為助催化劑(如鐵)能在費米能級上有效降低CNT/PdFe/CN雙金屬催化劑中鈀原子的3d電子密度,從而防止鈀氧化物的形成。因此,雙金屬鈀鐵納米粒子中的雙金屬協同效應被認為抑制了鈀的氧化,并有助于CNT/PdFe/CN催化劑的活性。
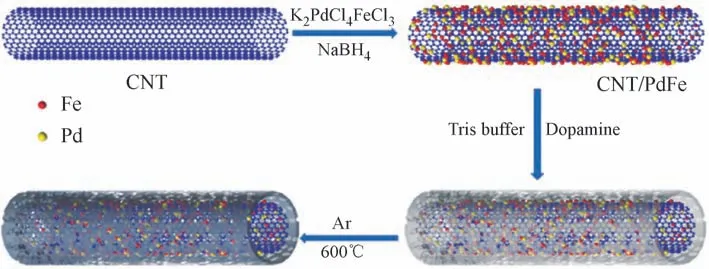
圖3 CNT/PdFe/NC催化劑的制備[12]
1.4 金屬離子/有機配體改性
已知第二金屬以原子形式引入到催化劑中可以提高活性,研究者還探索了第二金屬以離子或配合物形式引入后對催化劑的影響。Xu等[13]探究了Sn4+改性的PVP-Pd/Al2O3催化劑對p-CNB加氫的影響,催化劑活性改善,脫鹵被抑制。可能是Sn4+與—NO2基團中氧的作用增加了N==O鍵的極性,有利于活化的氫攻擊N==O鍵;同時,N==O鍵極性的增加使氯原子上更多p軌道孤對電子轉移到苯環上,從而使C—Cl鍵的鍵能增強,不易脫氯,見圖4。
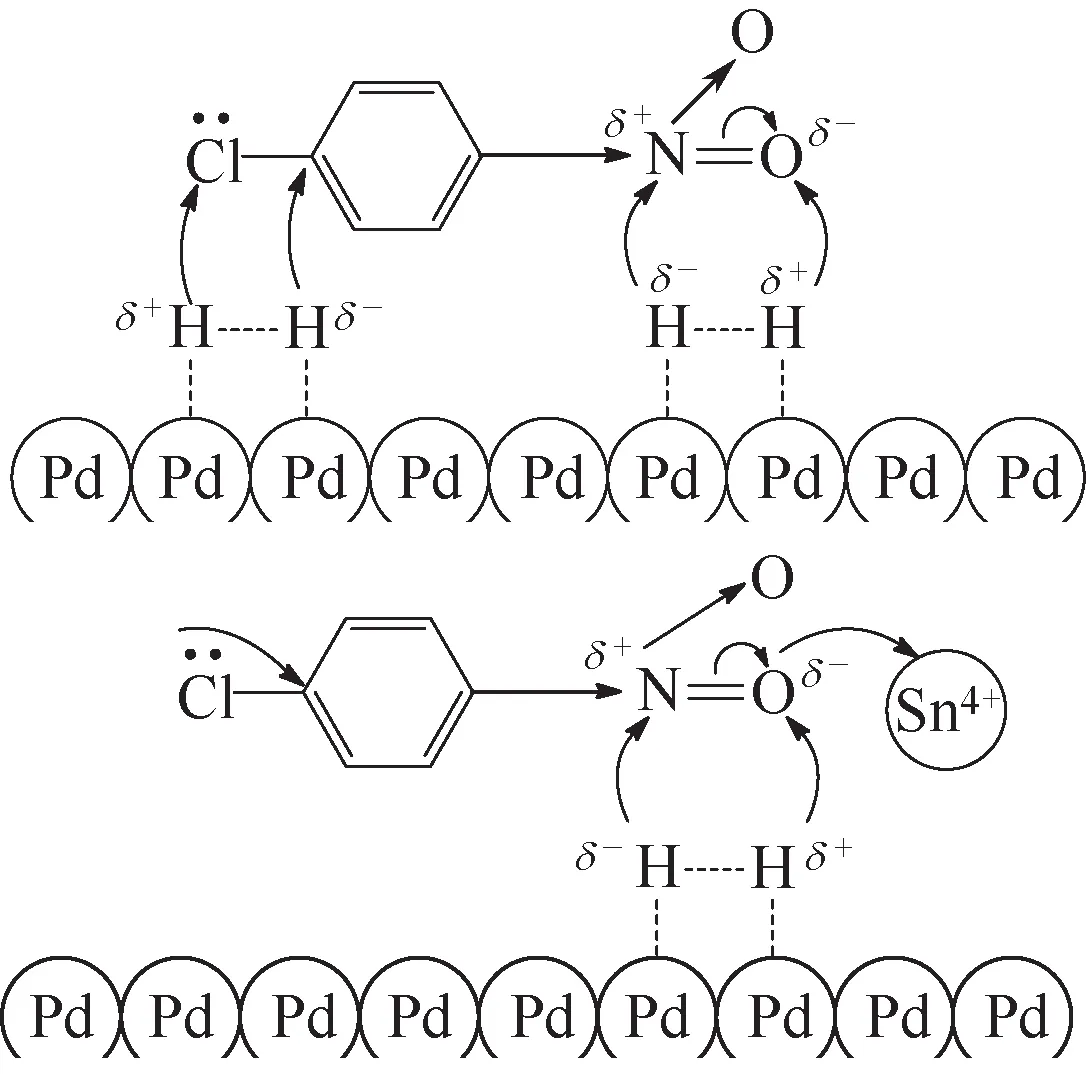
圖4 Sn4+對p-CNB加氫的改性機理[13]
Yu等[14]研究了金屬陽離子的影響。Fe3+使Pt/PS催化劑活性降低,Co2+使其活性增加,選擇性基本不變;Ni2+對催化劑活性和選擇性均有促進作用,Pt/PS-NiCl2催化劑可使主產物選擇性達到93.5%。作者猜測從鉑轉移到金屬陽離子上的電子可增強—NO2基團吸附到鉑表面的能力,吸附的金屬陽離子激活了極性的N==O鍵,從而提高了反應活性和選擇性。
Westerhaus等[15]報道了通過熱解非揮發性有機金屬胺絡合物制備活性氧化鈷/碳-氮催化劑。分析結果表明對于不同的有機配體,氧化鈷的粒度變化很大,較大的金屬顆粒催化性能更優。作者還探究了有機配體中氮的作用,表征結果顯示最低的結合能歸因于與金屬離子結合的吡啶型氮。總之,根據所用的有機配體,會產生不同的表面改性,進而控制反應的活性和選擇性。其中活性最高的是催化劑是通過將乙酸鈷(Ⅱ)-鄰菲羅啉絡合物吸附到碳載體上并熱解而得到的,以p-CNB、m-CNB為底物時,收率均可達95%。
1.5 構建納米管催化劑
除了調變催化劑組成來提高其性能,研究者還嘗試了構建具有特殊結構的催化劑(如納米管、納米棒催化劑)。
納米管狀材料催化是近年來最活躍的研究領域之一。Zhu等[16]探究了非晶態金屬-硼納米管(Co-B、Ni-B和Fe-B納米管)的催化性能,其中Co-B納米管對CNB加氫的催化活性較好,收率可達93.8%。其作者使用溶致非離子-陰離子混合表面活性劑液晶作為模板,合成了具有均勻尺寸的金屬硼納米管,為制備具有良好催化性能的金屬硼納米管提供了一條有效途徑。
Mo等[17]合成了三組分非晶態NiPB納米管,其直徑可控,在非晶態結構中比其雙組分對應物,如NiB合金具有更高的穩定性,且在對CNB加氫中主產物選擇性可達近100%。納米管較高的催化活性一方面歸因于其較大的表面積;另一方面,由于納米管內表面的負曲率,吸附在靠近孔壁的接觸層上的分子,由于與孔中所有原子的聯合相互作用而有較大的吸引勢能,該“限制效應”增加了分子與管壁上活性中心的碰撞概率。
Liu等[18]采用化學還原法制備了一系列NiPB催化劑。由于磷和硼的鈍化作用,NiPB催化劑比Raney Ni更穩定;而且較高含量的硼和磷使鎳活性中心變得更加高度不飽和,有利于提高加氫活性。在393K、1.2MPa的反應條件下,主產物p-CAN的選擇性大于99%。
2 均相催化劑
相較于多相催化劑,均相催化劑由于不存在內外擴散效應,且分散度高,所以反應效率高。但均相催化劑的回收比較困難。
2.1 金屬配合物催化劑
Jia等[19]報道了一種合成鈀(Ⅱ)-亞苯基橋聯的雙硫酮配合物的方法,并將該配合物用于硝基芳烴的催化還原。他們先采用一鍋法合成了亞苯基橋聯的雙硫酮配體(圖5),然后加入氯化鈀與其反應得到鈀(Ⅱ)-亞苯基橋聯的雙硫酮配合物,見圖6。將所得配合物用于硝基芳烴的催化還原時,在不脫氯的情況下,產率可達到98%。
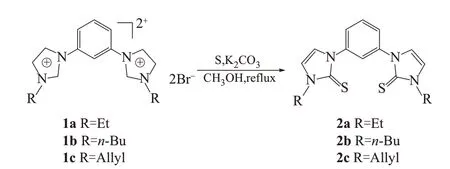
圖5 亞苯基橋聯雙(硫酮)化合物(2a~2c)的合成[19]
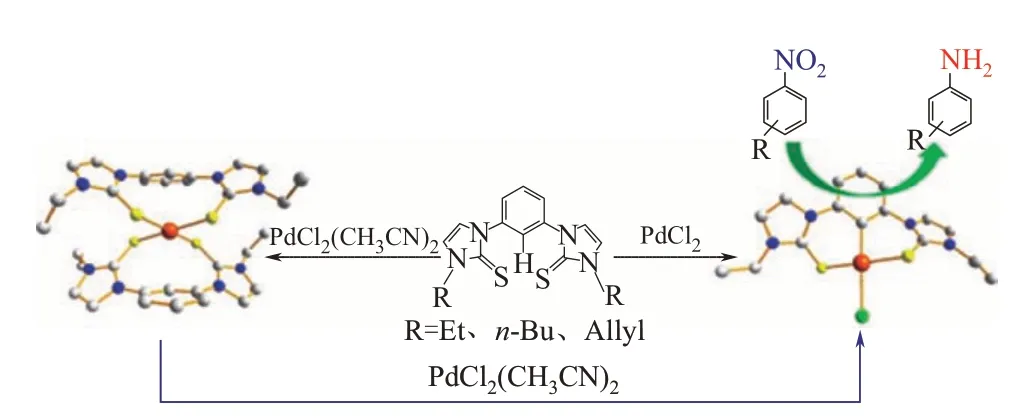
圖6 鈀(Ⅱ)配合物的合成與應用[19]
Lara等[20]報道了用氮雜卡賓(N-heterocyclic carbene,NHC)配體穩定的鉑納米顆粒催化劑(見圖7),催化實驗結果表明,通過選擇適當的NHC配體和調整NHC/Pt比,可在硝基加氫反應中表現出高活性和選擇性。作者認為由于IiPr2Me2配體中的芳香環通過π效應或元結效應與鉑顆粒表面相互作用,控制金屬顆粒的形態和尺寸,從而增強其穩定性。此外,NHC/Pt比還影響催化劑的活性,其中PtIPr0.2的催化活性最高,以p-CNB為底物時,收率可達95%。
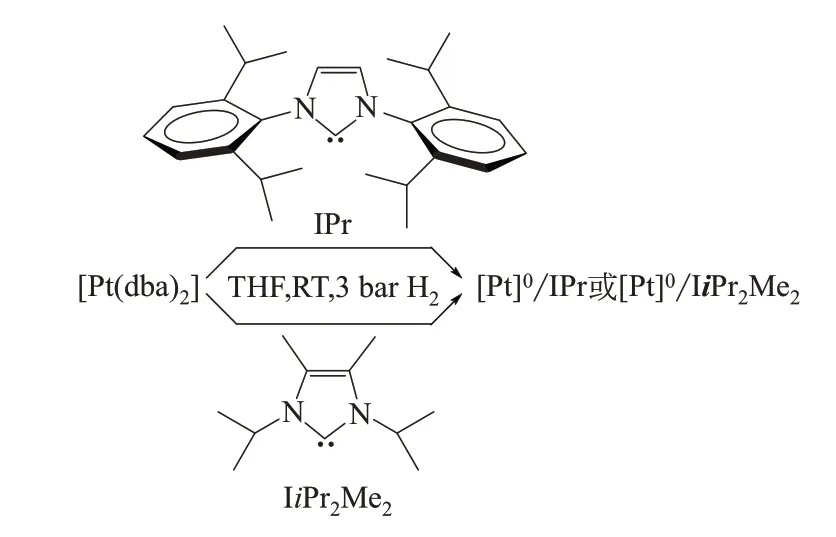
圖7 NHC配體穩定的鉑納米顆粒催化劑[20]
Wienhf?fer等[21]以鐵為活性中心,分別以三[(2-二苯基膦基)苯基]-膦、三[(2-二苯基膦基)乙基]-膦為配體合成鐵-膦配合物,其中,以三[(2-二苯基膦基)苯基]-膦為配體合成的鐵-膦配合物穩定性最高,且能高效催化各種硝基芳烴為相應的胺。在20bar H2(1bar=0.1MPa)、120℃的條件下催化p-CNB加氫,轉化率可達99%。
2.2 聚合物穩定的膠體金屬催化劑
聚合物穩定的金屬膠體由于其高比表面積和特殊的催化環境,表現出獨特的催化性能。聚合物不僅可以作為超細顆粒的穩定劑,還可以作為功能化超細顆粒的材料。
Tu等[22]制備了聚乙烯吡咯烷酮穩定的鉑膠體催化劑PVP-Pt用于催化還原CNB,并探究了金屬離子、金屬配合物對催化劑活性和選擇性的影響。Ni2+的引入對提高催化劑性能的效果最佳,而Mn2+則會導致不利影響。金屬聯吡啶配合物對催化劑性能影響較小,而金屬乙二胺配合物的影響則較顯著。8-羥基喹啉(8-HQ)是典型的螯合配體,金屬8-HQ配合物可提高選擇性,加入Fe(8-HQ)2+后,p-CAN的選擇性可達99.9%。作者認為金屬配合物對CNB的選擇性氫化有不同于金屬中心離子或配體的作用,金屬配合物的行為與金屬中心離子配位的配體數量有關,其與PVP-Pt催化劑的相互作用可改變表面鉑原子的電子密度,從而改變催化劑性能。
Yu等[23]報道了硫醇封端的聚(N-異丙基丙烯酰胺)穩定的膠體鉑催化劑對鹵代硝基苯選擇性加氫合成鹵代苯胺。作者采用可逆加成斷裂鏈轉移法(RAFT)制備了巰基封端的聚(N-異丙基丙烯酰胺)(見圖8),該聚合物既可穩定鉑納米顆粒,還可通過硫醇基團與鉑納米顆粒不飽和表面間的強相互作用,抑制脫鹵產物的生成。作者認為所得的高選擇性主要歸因于聚合物末端的巰基使鉑催化活性位點適度中毒,此外,被聚合物修飾的鉑表面的空間位阻效應避免了硝基還原過程中C—Cl鍵暴露于活性部位。
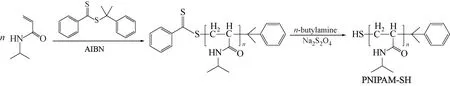
圖8 巰基封端的聚(N-異丙基丙烯酰胺)的合成[23]
3 催化反應體系的優化
通過優化催化反應體系可提高反應的選擇性,采用的優化措施主要有使用添加劑、改變反應介質等。
3.1 添加劑的影響
由于CNB選擇性加氫成CAN過程中易發生廣泛脫氯,因此可在反應體系中添加脫氯抑制劑來提高收率。常用的脫氯抑制劑有堿或其他給電子化合物,它們與金屬顆粒的相互作用提高了催化劑的電子性質,可有效抑制脫氯。
在使用雷尼鎳作為催化劑時,單紹軍等[24]分別使用乙醇胺、三乙烯四胺、環己胺和雙氰胺作為脫氯抑制劑,發現均能抑制脫氯。其中,雙氰胺效果最好,可完全抑制脫氯,產率可達到92.3%。還有研究者使用甲脒鹽作為抑制劑。另外,在使用Pt/C催化劑時,硫脲、乙醇胺等有機堿也可抑制脫氯,Han等[25]認為乙二胺中的氨基與鉑有很強的配位作用,降低了催化劑的活性中心,從而抑制C—Cl鍵的氫解。
部分有機硫化物能夠提高負載型貴金屬催化劑在加氫反應中的選擇性,Zhang等[26]在Pd/C催化劑中加入二苯硫醚,使p-CAN的選擇性達到99.6%。Makosch等[27]通過將有機硫醇與Pt/TiO2催化劑混合來改性,使硝基芳族化合物還原為對應的胺類產物的收率可達近100%,且發現改性劑結構影響選擇性:含有極性基團的改性劑(硫代甘油、1,6-己二硫醇和α-硫辛酸)可使主產物選擇性達到近100%,而非極性改性劑(1-十二烷硫醇)則次之。
另外,無機堿也常用作催化加氫脫氯抑制劑。Han等[25]使用PVP-Pt催化劑時,在反應體系中加入適量NaOH,可將p-CAN的選擇性從78.5%提高到89.9%。他們還發現添加KOH、K2CO3、NaOAC等均能提高p-CNB的選擇性。堿可作為活性金屬中心的配體參與催化,且堿可以中和反應過程中產生的鹽酸,從而促進反應。
在反應體系中加入脫氯抑制劑有利于提高主產物的選擇性,但部分添加劑會使催化劑局部中毒而降低催化劑活性;此外,還需要額外的步驟除去添加劑,使反應過程復雜化。
3.2 溶劑的影響
溶劑對催化加氫過程有顯著影響,其中包括氫的溶解度、溶劑的相互作用等,目前常用的有機溶劑如甲醇、己烷、環己烷、乙酸乙酯、乙醚和四氫呋喃常用于液體和常規氣相氫化。其中部分有機溶劑易揮發、有毒,并伴隨著嚴重的健康危害和環境問題。因此,替換掉有毒溶劑對實現綠色生產具有重要意義。
離子液體是一類熔點較低(<100℃)的熔鹽,具有蒸汽壓極低、極性可調和潛在的易回收性等特征,是替代有毒有機溶劑的理想溶劑之一。Xiao等[28]使用離子液體[BMIM][BF4]作為離子液體類共聚物穩定的鉑納米團簇催化劑的反應介質,通過o-CNB加氫來考察其性能,實驗結果顯示離子液體[BMIM][BF4]不僅有利于產品分離和催化劑再循環,而且可提高選擇性,o-CAN的選擇性可達96%。
由于離子液體的合成和保存較困難,超臨界二氧化碳(scCO2)可成為更綠色經濟的替代品。scCO2具有不易燃、相對惰性、易分離等優點,此外,它對反應速率和產物選擇性有積極影響。Meng等[29]以Ni/TiO2為催化劑,在35℃超臨界二氧化碳、乙醇和正己烷中,研究了CNB催化加氫反應。以超臨界二氧化碳為反應介質時,CAN選擇性可高達99.5%;而在乙醇和正己烷中,無法實現對CAN的高選擇性。Ichikawa等[30]以scCO2為反應介質時,發現CNB在Pt/C催化劑上的脫氯被顯著抑制,在100%的轉化率下,對CAN的選擇性可大于99%。但目前超臨界應用于CNB選擇性催化加氫的研究還較少,對于其穩定性和作用機理尚不明確,仍需要進一步探索。
4 結語
CNB選擇性加氫制備CAN的兩類催化體系的改進方法及其特點如表1所示。

表1 CNB選擇性加氫催化劑的改進方案與特點
CNB選擇性加氫催化劑應同時滿足高活性和抑制脫氯的高選擇性要求。對于均相催化劑,用配合物/聚合物來穩定活性金屬是非常有效的策略,添加金屬助劑可有效提高反應效率,但在穩定性、制備的簡單性和循環使用方面仍有待提升。相較于均相催化劑,多相催化劑在工業上有著更廣泛的應用,其制備工藝簡單、易重復利用、成本較低。當前,多相催化劑的性能優化研究仍任重道遠,應特別重視其金屬相和載體的改進,對于金屬相:調節金屬分散度可改變電荷密度和電化學結合能,促進底物活化;引入第二種金屬形成合金催化劑,或者引入金屬離子/配合物助劑,可改善—NO2基團的吸附并增加N==O鍵的極性,同時增強C—Cl鍵的鍵能,達到提高催化活性和選擇性的目的。對于載體,通過調節其孔結構和比表面積,改善活性組分分散度,也有利于增強機械性能;通過調變其表面性質,構建缺陷位或者摻雜雜原子來錨定活性位點;通過載體上的給電子基團來抑制—NH2基團的給電子能力,從而抑制脫氯;利用載體表面電子遷移形成的空穴來激活N==O鍵,提高反應速率。特殊結構的催化劑,如金屬硼納米管等,是性能優異多相催化劑。單原子催化劑的活性好,但其穩定性仍是亟待解決的問題。在這些基礎上,構建具有耐性優、穩定性好、選擇性高的多相催化劑具有重要的理論與實際意義。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
