酶預處理對木質纖維素納米纖絲性能的影響
2021-08-13 09:51:32賀禮龍,黃璐瑤,高文花,曾勁松,陳克復
大連工業大學學報 2021年4期
賀 禮 龍, 黃 璐 瑤, 高 文 花, 曾 勁 松, 陳 克 復
( 華南理工大學 制漿造紙工程國家重點實驗室, 廣東 廣州 510640 )
0 引 言
納米纖維素纖絲(CNF)是通過解構植物纖維素纖維而獲得的納米級材料。CNF長度為幾百納米到幾微米,直徑為5~100 nm。CNF具有高比表面積、高機械強度、低密度、可生物降解性和生物相容性等優越性能[1],已成為纖維素科學研究領域的熱點。
通常在生產CNF之前,需去除原材料中的木質素,這必將導致大量的能量和化學品消耗,對環境的影響比較大。相對于納米纖維素纖絲,木質纖維素納米纖絲(LCNF)制備成本低,過程污染小[2],其制備方法與CNF幾乎一致。目前,有許多方法可用于生產CNF,如機械精制、冷凍破碎、超聲處理和微波處理等,其中機械精制工藝已普遍應用于生產,如高壓均質化、微流化和研磨化。但是,精制過程中的高能量輸入以及精制后產品的異質性強烈限制了其應用[3-4]。為了克服這些挑戰,在機械精制之前,可采用化學/生物預處理來“打開/松散/軟化”纖維素原材料。盡管各種基于化學的預處理方法都可以實現這一目標,如TEMPO氧化、羧甲基化、磺化等,但是這些化學過程會產生一定的污染物,具有相當大的環境風險,限制了產品的進一步規模化推廣應用[5-6]。與化學預處理方法相比,生物處理因其酶負荷低、酶選擇性高、反應條件溫和而被認為是更具吸引力的方法。
本研究采用內切葡聚糖酶和木聚糖酶進行預處理來制備LCNF,分析了預處理前后組分的變化,并探討其對LCNF保水值、結晶度、比表面積等性能的影響,以期為高性能LCNF的綠色低能耗制備提供理論基礎。
1 實 驗
1.1 材料與儀器
材料:漂白化學熱磨機械漿(BCTMP),芬歐匯川(常熟)紙業有限公司,游離度300 mL,白度79.0% ISO;商業內切葡聚糖酶(Banzyme 2900),芬歐匯川有限公司;木聚糖酶,麥克林生化科技有限公司;內切葡聚糖酶和木聚糖酶的酶活性分別為7.3和327 IU/g。亞氯酸鈉、冰醋酸、丙酮;分析純。
儀器:MKCA6-2J超微粒研磨儀,日本增幸產業株式會社;DionexICS-5000離子色譜儀,賽默飛世爾科技(中國)有限公司;Multimode-8原子力顯微鏡,德國Bruker公司;UV-1900紫外分光光度計,日本島津儀器有限公司;PCD-03粒子電荷探測儀,德國Mütek公司。
1.2 實驗方法
1.2.1 原料預處理
將紙漿浸泡在去離子水中12 h,再采用實驗室規模的Valley打漿機對纖維進行疏解處理。處理后的紙漿離心脫水并置于4 ℃平衡水分。
1.2.2 漿料脫木質素處理
漿料木質素脫除采用改進的測定綜纖維素含量的方法(GB/T 2677.10—1995)。將平衡水分后的20.0 g漿料與650 mL去離子水、5 mL冰醋酸以及6.0 g亞氯酸鈉充分混合,移入1 000 mL錐形瓶中,搖勻,扣上25 mL錐形瓶,置于75 ℃恒溫水浴中加熱2 h,反應1 h后加入一次冰醋酸和亞氯酸鈉。定期搖動錐形瓶以確保試劑完全反應。反應結束后,將漿料放入冰水浴中冷卻,并用去離子水反復洗滌至濾液不呈酸性為止,再用丙酮洗滌3次,收集漿料并置于4 ℃平衡水分。
1.2.3 酶預處理
將漿料置于50 ℃和200 r/min的條件下在50 mmol/L檸檬酸-檸檬酸鈉緩沖液(pH 5.2)中進行酶預處理,底物質量分數為5.0%。預處理期間內切葡聚糖酶的用量為5 mg/g纖維底物,反應時間為1.5 h。反應結束后,將漿料置于95 ℃ 的水浴中10 min使酶滅活,離心收集酶解液用于后續檢測分析。酶預處理后的漿料用去離子水洗滌至中性并置于4 ℃平衡水分。與內切葡聚糖酶相比,木聚糖酶的用量為5 IU/g纖維底物,反應時間為0.5 h,其他操作同內切葡聚糖酶的處理。內切葡聚糖酶和木聚糖酶預處理的漿料分別命名為EP和XP,未進行酶預處理的漿料命名為BP。
1.2.4 機械納米原纖化處理
分別將酶預處理和未經酶預處理的漿料稀釋至質量分數為1.0%,經超微粒研磨儀進行研磨制備LCNF。具體工藝:在磨盤間隙為0和-20 μm 下依次研磨5次,轉速為1 500 r/min;磨盤間隙調至-50和-80 μm,依次研磨10次,轉速為2 000 r/min;磨盤間隙再調至-100 μm研磨15次,轉速為2 000 r/min。得到內切葡聚糖酶/機械處理、木聚糖酶/機械處理和純機械處理的懸浮液分別命名為E-LCNF、X-LCNF和N-LCNF。
1.2.5 測試與表征
1.2.5.1 組分分析
BP、EP和XP樣品的木質素含量及酶解液中糖分組成,根據改進文獻[7]的分析方法進行。
稱取0.3 g干燥至恒重的樣品,在30 ℃用3 mL 質量分數72%的H2SO4水解1 h,每10 min 攪動一次,使其水解反應均勻。反應結束后,將反應混合物轉移到耐壓瓶中,加入84 mL去離子水,并在121 ℃下高壓保溫1 h。將反應后的液體過濾,收集酸水解溶液,使用紫外-可見分光光度計從205 nm處的吸光度分析計算酸溶性木質素的含量。將剩余的固體殘留物在105 ℃干燥至恒重,記錄干燥固體的質量,并用于計算酸不溶性木質素的含量。
取9.3 mL去離子水加入耐壓瓶中,再加入0.7 mL質量分數72%的H2SO4,最后加入10 mL 收集的酶解液,溶液在121 ℃高壓保溫1 h。將酶解液和酸解后的酶解液分別通過離子色譜系統分析單糖組分。
1.2.5.2 紅外光譜分析
通過傅里葉變換紅外光譜分析BP、EP和XP樣品的氫鍵特征參數。使用Peak-Fit軟件結合高斯分布函數研究吸收光譜(3 700~3 000 cm-1)以分析氫鍵含量、能量和不同氫鍵模型的鍵長。
EH=(ν0-ν)/Kν0
(1)
式中:EH為氫鍵能量,kJ/mol;ν0為標準游離羥基頻率,3 650 cm-1;ν為樣品的羥基頻率,cm-1;K為常數,6.7×10-2kJ-1。
(2)
式中:R為鍵長,10-10m;ν0為標準游離羥基的拉伸振動頻率,3 600 cm-1;ν為樣品的羥基頻率,cm-1。
1.2.5.3 形貌分析
將LCNF配制成質量分數為10-6~10-5的懸浮液,15 ℃超聲分散5 min,取一滴超聲分散好的溶液滴在云母片上,自然風干后用原子力顯微鏡觀察其形態特征。采用Nano Measurer 1.2軟件統計LCNF的直徑分布。
1.2.5.4 保水值
保水值反映了纖維孔隙中的總水分,該水分與總孔隙體積密切相關,是纖維素原纖維吸水和溶脹能力或原纖化程度的量度。采用改進的TAPPI T256方法[8]進行測量,質量分數為1.0%的LCNF懸浮液在3 000g的離心力下離心30 min,離心后的樣品稱重,105 ℃干燥24 h至恒重。保水值由離心后底物中水分的質量與絕干LCNF質量的比值計算得出。
保水值=[(m1-m2)/m2]×100%
(3)
式中:m1和m2分別為離心后底物的質量和恒重后底物的質量,g。
1.2.5.5 X射線衍射測試
LCNF的結晶度通過X射線衍射儀使用Cu-Kα輻射確定。在40 kV、30 mA以2.0°/min在5°~60°內掃描樣品。
結晶度指數=[(I200-Iam)/Iam]×100%
(4)
式中:I200為22.5°附近2θ處200晶格平面的衍射強度;Iam為18°附近2θ處非結晶區的衍射強度。
Dhkl=kλ/βcosθ
(5)
式中:Dhkl為晶體在200晶格平面上的平均微晶尺寸,nm;k為Scherrer常數,0.89;λ為Cu-Kα輻射的波長,0.154 18 nm;β為200晶格平面半峰高的寬度,rad;θ為布拉格角度,rad。
1.2.5.6 比表面積
LCNF樣品的比表面積采用文獻[9]中剛果紅染料吸附法測定。用剛果紅(底物質量分數5%~30%)處理質量分數0.2%的LCNF水性懸浮液,60 ℃振蕩24 h,并在5 000 r/min離心20 min。用紫外-可見分光光度計在495 nm處檢測上清液的吸光度,根據剛果紅標準曲線計算上清液中的剛果紅濃度。
Acell=6.024×1023m1Ad/Mr
(6)
式中:Acell為比表面積,m2/g;m1為纖維吸附的飽和值,g/kg;Ad為染料分子的面積,1.73 nm2;Mr為染料分子的相對分子質量,696.68。
1.2.5.7 懸浮液特性
LCNF樣品的表面電荷密度通過粒子電荷探測儀進行測量,標準滴定液為Poly-DADMAC。Zeta電位通過流動電位法Zeta電位儀進行測定。
2 結果與討論
2.1 酶預處理對纖維結合力的影響
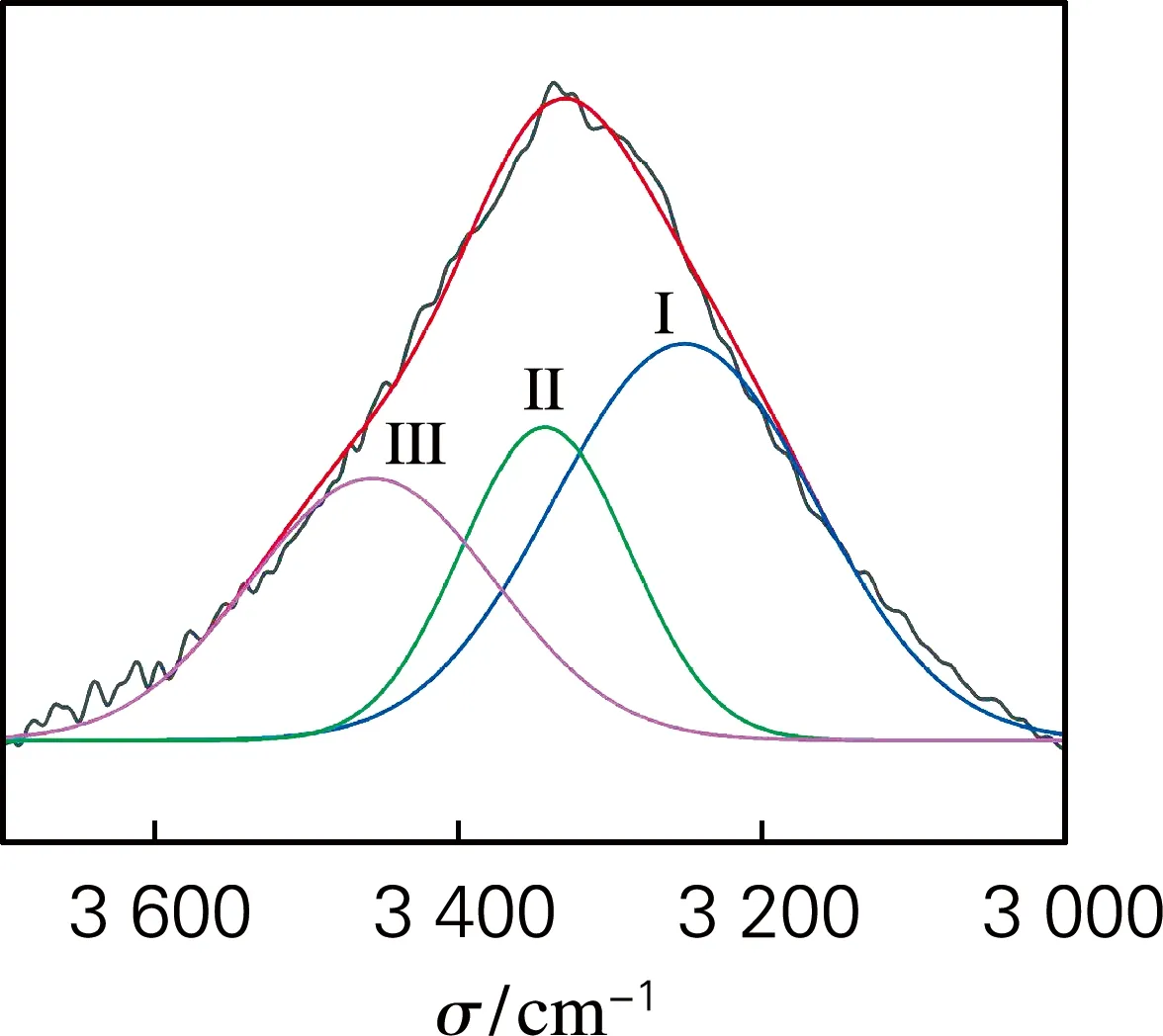
根據紅外光譜吸收頻率偏移程度以及譜圖的面積變化評價酶預處理對漿料纖維氫鍵特性的影響。分子間和分子內氫鍵的伸縮振動特征峰位于3 000~3 700 cm-1區域。其中,代表O(2)H…O(6)和O(3)H…O(5)的分子內氫鍵及代表O(6)H…O(3′)的分子間氫鍵的特征吸收波數分別在3 455~3 410 cm-1、3 375~3 340 cm-1和3 310~3 230 cm-1。氫鍵譜帶的二階導數可用于確定不同氫鍵模型的波數,對圖1中吸收峰氫鍵區域進行分峰擬合,分析不同類型氫鍵的特征,得到紅外分峰擬合圖譜(圖2)。
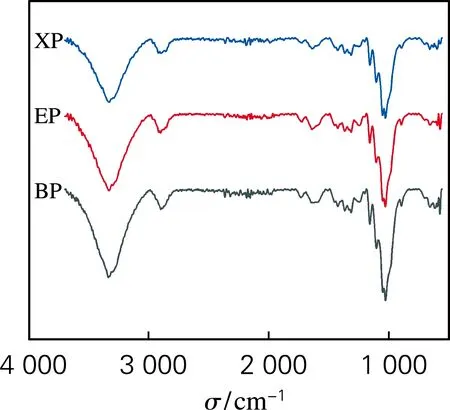
圖1 BP、EP與XP的紅外光譜
(a) BP
如表1所示,所有分峰擬合的R2均高于0.98,這表明不同氫鍵模型的擬合結果能夠充分反映不同氫鍵模型的信息。結果表明,酶預處理后,纖維分子間與分子內氫鍵含量發生變化,纖維的氫鍵逐漸發生“重組”現象,說明酶預處理可以疏松纖維的結構,有利于后續納米原纖化。
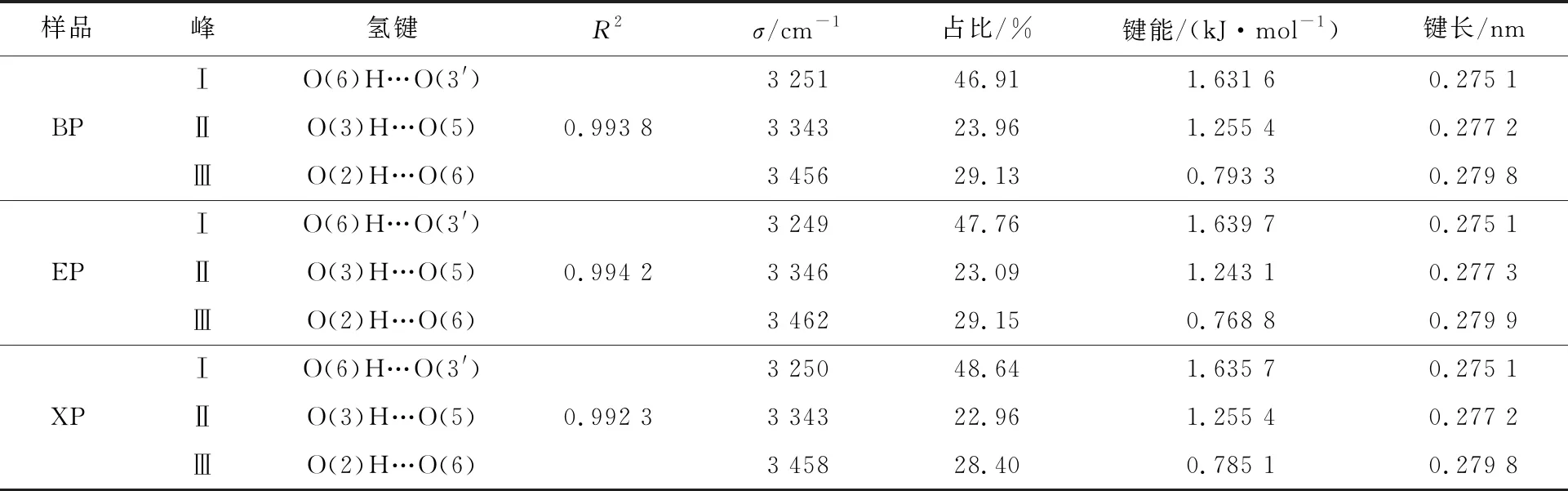
表1 酶預處理樣品紅外圖譜氫鍵區域的分峰擬合
2.2 酶預處理對漿料纖維組分的影響
酶預處理過程可以減弱微纖絲之間的氫鍵作用力,表1結果已得到證實。酶解液中單糖及低聚糖的含量可以反映酶預處理過程對纖維復雜多層壁結構的改變,同時也進一步證實酶預處理過程可以疏松木質纖維,其中,酶解液中低聚糖由酶解液酸解后單糖的含量和組成間接反映。
經內切葡聚糖酶和木聚糖酶預處理后的漿料得率分別為92.25%和83.22%,酶解前后漿料主要化學組分組成如表2所示。由表2可以看出,內切葡聚糖酶預處理后,酶解液糖含量和酶解液酸解后糖含量都很少,說明預處理過程主要以疏松纖維為主,水解的漿料纖維較少。這是可能由于內切葡聚糖酶在纖維的非結晶區域隨機水解β-1,4-葡萄糖苷鍵,使纖維素的非結晶區域變疏松,并較少地將纖維素或半纖維素降解為單糖或

表2 酶解液酸解前后單糖的質量分數
可溶性低聚糖。與內切葡聚糖酶處理的樣品相比,經木聚糖酶處理后,酶解液及酶解液酸解液中的木糖含量明顯增高。這是由于木聚糖酶能選擇性地降解半纖維素中的木聚糖為單糖或可溶性低聚木糖。但該水解大多發生在微纖維之間的半纖維素中,而基層纖維致密結構之間的半纖維素卻很難被直接接觸或水解[10]。此外,內切葡聚糖酶處理的樣品中木質素質量分數為11.21%,與未經酶預處理的樣品(11.51%)相差無幾,而木聚糖酶處理的樣品木質素含量有所下降,其質量分數為6.08%。這是由于在植物纖維中,半纖維素與木質素之間存在化學鍵,構成木質素-碳水化合物復合體(LCC)[11]。因此,木聚糖酶處理在去除半纖維素成分的同時也會使一部分木質素脫落,并清楚地暴露細胞壁的原纖維網絡。
2.3 酶預處理對LCNF形貌與尺寸的影響
由圖3可以看出,未經酶預處理的N-LCNF呈現一種樹形結構,且“樹干”與“樹枝”的尺寸差別較大[9],從直方圖中能夠直觀地看到該樣品的直徑主要分布在20~60 nm,尺寸分布跨度大,相對分散。經過酶預處理的E-LCNF和X-LCNF都呈現相互交織的無規則網絡結構。E-LCNF的直徑主要分布在15~35 nm,分布相對集中,由于內切葡聚糖酶有效疏松纖維結構,機械研磨能夠使纖維剝離出更細小的纖絲;X-LCNF直徑主要分布在20~50 nm,分布相對較為集中,因為木聚糖酶能夠降解半纖維素,破壞纖維的固有抗降解屏障性,打開纖維網絡,有助于纖維納米原纖化。由于EP的木質素含量高于XP,木質素是一種已知的抗氧化劑,它能穩定機械處理過程中形成的纖維素自由基。纖維素自由基具有極強的反應性,可以參與復合反應(交聯),在木質素含量較低的情況下會不利于原纖化,在木質素含量較高的情況下,木質素清除自由基的能力導致纖維素的交聯不那么明顯,這使得纖維可以更好地解構[20],因此E-LCNF的直徑小于X-LCNF。
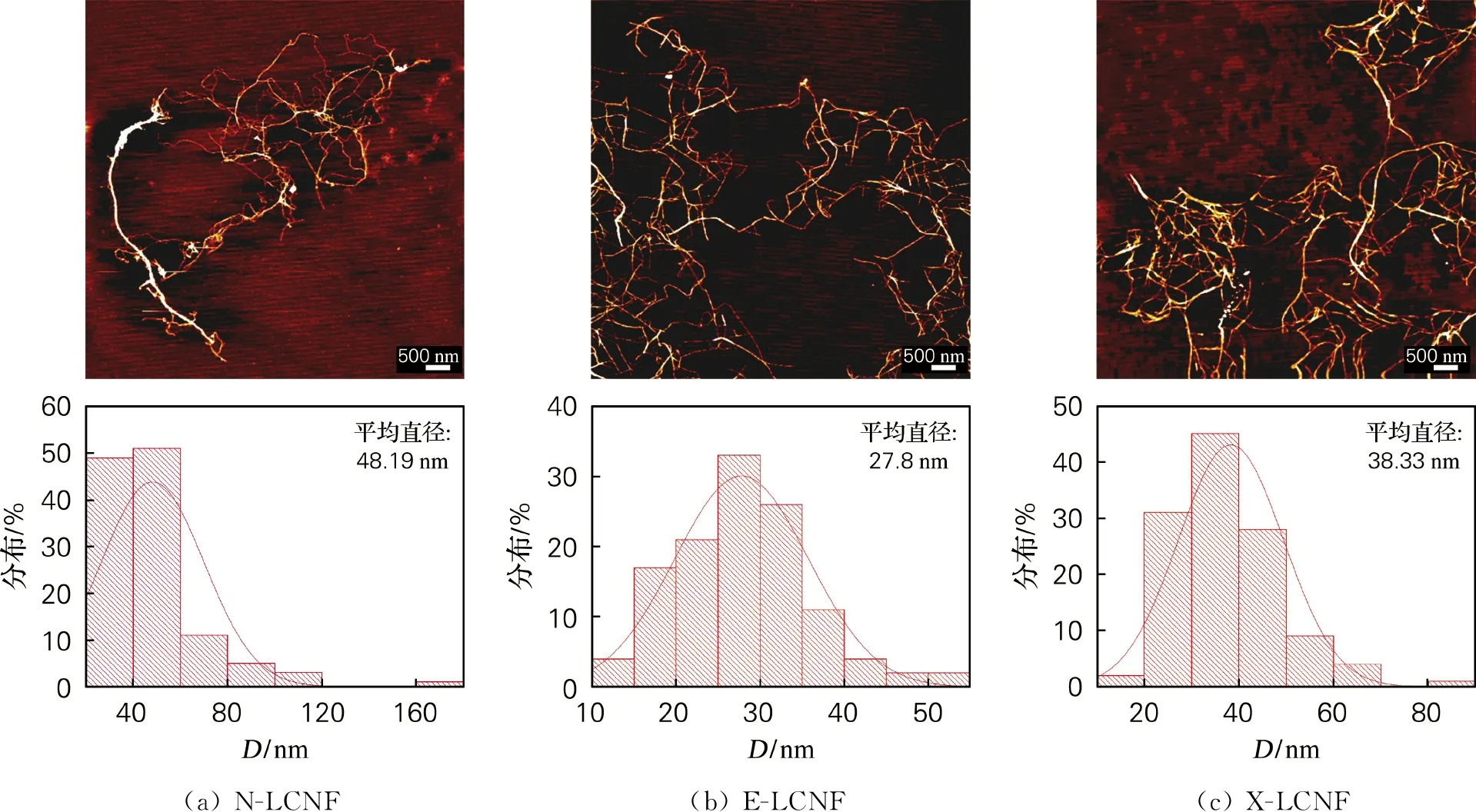
圖3 LCNF的AFM形貌結構及其直徑分布
2.4 酶預處理對LCNF性能的影響
如表3所示,E-LCNF樣品獲得了最高的保水值564%,表明該樣品在內切葡聚糖酶/機械研磨處理后發生顯著的原纖化,纖維裸露的羥基增加,結合水分子能力增強。由于內切葡聚糖酶可以高效率和選擇性地水解纖維中的非結晶區,從而使樣品產生較小的纖維碎片和增加纖維素纖維在水中的溶脹度。此外,內切葡聚糖酶可能會破壞微纖維之間的氫鍵連接[12-13],這對從纖維表面解離出纖絲具有積極影響,也意味著可以在纖維上建立更多的水可滲透的空腔[14],從而導致保水值的增加,同時也提高了樣品的比表面積。

表3 酶預處理制備所得LCNF的性能
X-LCNF的保水值顯著提高,表明木聚糖酶預處理有助于“打開/松散/軟化”纖維素原材料。半纖維素組合物采取高度分支的多態多糖的形式,通過氫鍵結合相鄰的纖維素微原纖維來增強細胞壁強度,但其分支數量和組成的變化可以顯著改變細胞壁強度和微觀結構[15]。木聚糖酶可以使紙漿纖維中的半纖維素結構自然地解構,有益于纖維原纖化和微原纖或納米原纖的形成[16]。
因此,木聚糖酶預處理可以破壞纖維羥基之間的分子間和分子內氫鍵,隨后的機械原纖化增加了原纖維的內表面和外表面面積,使水分子更容易滲透到原纖維之間以增加保水值。然而,由于木聚糖酶降解了部分半纖維素,半纖維素可以抑制纖維聚合,促進纖維形成開放的、多孔的結構[17-18],且半纖維素比纖維素具有更高的水可及性[19],因此X-LCNF的保水值稍低于E-LCNF。
纖維內部的結晶區和非結晶區之間存在相互干擾。從圖4中可以看出,經過酶預處理和未經酶預處理的樣品在XRD譜圖上相差不大,說明酶預處理過程中纖維內部結晶區域的晶型微觀結構變化不顯著。
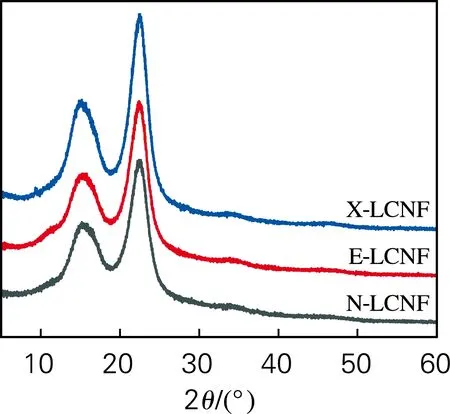
圖4 LCNF的X射線衍射譜圖
內切葡聚糖酶預處理樣品的結晶度和微晶尺寸略有增加。通過木聚糖酶預處理制備X-LCNF的結晶度增加較為明顯(表3),結晶纖維素被包埋在非結晶組分中,木聚糖酶處理后,附著在纖維表面的半纖維素被去除,提高了LCNF的結晶度。
如表3所示,E-LCNF懸浮液的Zeta電位最高,說明內切葡聚糖酶酶解結合機械研磨制備的木質纖維素納米纖絲有著良好的分散能力。這是由于內切葡聚糖酶能隨機切割纖維使其變短,并破壞氫鍵減弱纖維內部纖絲的作用力,在機械力的作用下更易剝離出直徑較小且尺寸分布均勻、比表面積大的纖維。此外,半纖維素表面帶有負電荷,能夠在纖維間產生排斥力[10],且側鏈基團也有助于納米纖維之間的空間排斥,因此半纖維素的存在促進了LCNF懸浮液的膠體穩定性。由于在預處理過程中木聚糖酶降解了部分半纖維素,靜電排斥的作用減弱,但同時木聚糖酶酶解打開了纖維網絡,在后續機械作用下原纖化程度較高,產生的納米纖維尺寸較為均勻,綜合作用下,X-LCNF的Zeta電位稍低于E-LCNF,但電荷密度最低。N-LCNF中雖然半纖維素含量高,能夠提供較強的靜電排斥力,但由于未經酶預處理,使得N-LCNF在機械研磨后的尺寸較大且分布不均勻,這可能導致靜電排斥力不足以抵抗其傾向于凝聚的趨勢,故而N-LCNF的Zeta電位在三者中最低。正如圖5所示,可以直觀地看到,N-LCNF懸浮液靜置后會絮聚沉降,分散性較差,而E-LCNF與X-LCNF由于Zeta電位絕對值都大于40 mV,表現出良好的分散性。

圖5 靜置5 d的LCNF分散液
3 結 論
采用酶預處理結合機械研磨的方法制備了不同特性的LCNF樣品,探究了酶預處理過程對LCNF性能的影響。結果表明,內切葡聚糖酶在預處理過程中對漿料的組分改變較為細微,得率高,隨機水解非結晶區纖維疏松了木質纖維微觀結構,同時也減弱了纖維間的作用力,在機械力剝離與切斷的作用下原纖化明顯,尺寸小且分布均勻。因此,E-LCNF的保水值、比表面積以及Zeta電位最高。木聚糖酶預處理時降解了半纖維素,同時也使部分木質素脫落,導致得率相對較低但軟化了纖維原料,納米原纖化后所得樣品X-LCNF 具有最高的結晶度,保水值、比表面積以及Zeta電位均只稍低于E-LCNF。機械法所得樣品N-LCNF由于未經預處理,其尺寸大且分布很不均勻,保水值、比表面積以及Zeta電位最低。總之,內切葡聚糖酶可以疏松木質纖維結構,進而促進木質纖維納米原纖化,提高LCNF性能;木聚糖酶可以降解半纖維素并脫落部分木質素,“打開/松散/軟化”了木質纖維原材料,改善了LCNF性能。
